Deep Research
Falcon
(Q72DT2-deep-research-falcon.md)
Falcon
(Q72DT2-deep-research-falcon.md)The research report should be a detailed narrative explaining the function, biological processes, and localization of the gene product. Citations should be given for all claims.
You should prioritize authoritative reviews and primary scientific literature when conducting research. You can supplement
this with annotations you find in gene/protein databases, but these can be outdated or inaccurate.
We are specifically interested in the primary function of the gene - for enzymes, what reaction is catalyzed, and what is the substrate specificity? For transporters, what is the substrate? For structural proteins or adapters, what is the broader structural role? For signaling molecules, what is the role in the pathway.
We are interested in where in or outside the cell the gene product carries out its function.
We are also interested in the signaling or biochemical pathways in which the gene functions. We are less interested in broad pleiotropic effects, except where these elucidate the precise role.
Include evidence where possible. We are interested in both experimental evidence as well as inference from structure, evolution, or bioinformatic analysis. Precise studies should be prioritized over high-throughput, where available.
Plan
- Objective 1 (verification): Confirm the identity of aprA (DVU_0847; UniProt Q72DT2) from Desulfovibrio vulgaris Hildenborough (DvH), its enzyme class, domains, complex membership (AprAB), and pathway context. (Completed using sources below.) (barton2014hydrogensulfidea pages 16-19, leavitt2019proteomicandisotopic pages 7-9, leavitt2019proteomicandisotopic pages 5-7)
- Objective 2 (recent literature): Prioritize 2023–2024 primary and review literature on APS reductase (AprAB) and the associated Qmo electron transfer system in sulfate reducers. (Completed using 2023 Frontiers RB‑TnSeq study and a 2024 scholarly chapter.) (d’ermo2024thecomplexinterplay pages 5-8, trotter2023largescalegeneticcharacterization pages 4-6)
- Objective 3 (organism-specific evidence): Extract DvH-specific locus, genetic/operon context, essentiality/fitness, proteomics expression, and interaction with Qmo. (Completed from DvH-focused proteomics and RB‑TnSeq datasets.) (leavitt2019proteomicandisotopic pages 7-9, trotter2023largescalegeneticcharacterization pages 4-6, leavitt2019proteomicandisotopic pages 5-7)
- Objective 4 (synthesis): Organize key concepts/definitions, reaction chemistry, cofactors, localization, pathway integration, real-world implications, expert viewpoints, and any available quantitative data. (Completed.) (barton2014hydrogensulfidea pages 16-19, d’ermo2024thecomplexinterplay pages 5-8, barton2014hydrogensulfidea pages 4-7)
- Objective 5 (final report): Write a comprehensive, fully cited research report with URLs and publication dates. (Completed.)
Comprehensive Research Report: aprA (DVU_0847; UniProt Q72DT2) of Desulfovibrio vulgaris Hildenborough
1) Key concepts and definitions with current understanding
- Target verification and identity: The gene aprA in DvH (ordered locus DVU_0847; UniProt Q72DT2) encodes the alpha subunit of adenylyl-sulfate (adenosine-5′-phosphosulfate, APS) reductase, the central enzyme in dissimilatory sulfate reduction that reduces APS to sulfite and AMP; it functions as a heterodimer with AprB (beta subunit) to form APS reductase (AprAB). In Desulfovibrio and other sulfate-reducing bacteria (SRB), AprAB is the canonical enzyme for this step in energy metabolism, distinguishing it from the assimilatory pathway variants. In DvH proteomics, AprA is explicitly mapped to DVU_0847, confirming the gene-protein assignment in this organism. (Barton et al., 2014; Leavitt et al., 2019) https://doi.org/10.1007/978-94-017-9269-1_10; https://doi.org/10.3389/fmicb.2019.00658 (barton2014hydrogensulfidea pages 16-19, leavitt2019proteomicandisotopic pages 7-9, leavitt2019proteomicandisotopic pages 5-7)
- Enzyme class and reaction: APS reductase (EC 1.8.99.2) catalyzes APS + 2e− + H+ → AMP + sulfite (SO3(2−)). The AprA subunit houses the catalytic FAD, while AprB contains iron-sulfur clusters that mediate electron transfer, consistent with a flavoprotein-iron–sulfur oxidoreductase mechanism. (Barton et al., 2014) https://doi.org/10.1007/978-94-017-9269-1_10 (barton2014hydrogensulfidea pages 16-19)
- Electron donor system and complex association: Electron delivery to AprAB is mediated by the membrane-associated Qmo (quinone-interacting membrane-bound oxidoreductase) complex (QmoABC), which couples quinone pool redox chemistry to APS reduction; genomic co-localization of aprAB with qmoABC is common in SRB and functionally linked to APS reduction. (Barton et al., 2014; D’Ermo et al., 2024; Saxena et al., 2023) https://doi.org/10.1007/978-94-017-9269-1_10; https://doi.org/10.1007/978-3-031-54306-7_15; https://doi.org/10.3389/fmicb.2023.1086021 (barton2014hydrogensulfidea pages 16-19, d’ermo2024thecomplexinterplay pages 5-8, saxena2023integrationoftext pages 5-6)
- Pathway context: In DvH’s dissimilatory sulfate reduction, sulfate is activated by ATP sulfurylase (Sat) to APS, reduced by AprAB to sulfite, and then by dissimilatory sulfite reductase (DsrAB) to sulfide, with multiple electron transfer modules (including Qmo and downstream complexes) supporting energy conservation. (Barton et al., 2014; D’Ermo et al., 2024) https://doi.org/10.1007/978-94-017-9269-1_10; https://doi.org/10.1007/978-3-031-54306-7_15 (barton2014hydrogensulfidea pages 16-19, d’ermo2024thecomplexinterplay pages 5-8)
2) Recent developments and latest research (priority to 2023–2024)
- Genome-wide fitness genetics in DvH (2023): A large RB‑TnSeq study in DvH mapped essential and conditionally essential genes across 2,741 loci under diverse conditions, providing a reference for sulfate-reduction gene fitness. Although focused broadly on metabolism, this dataset defines condition dependencies and essential gene sets relevant to sulfate reduction, establishing a contemporary genetic framework that includes the sulfate reduction module (sat/apr/dsr). (Trotter et al., 2023, Frontiers in Microbiology; Mar 2023) https://doi.org/10.3389/fmicb.2023.1095191 (trotter2023largescalegeneticcharacterization pages 4-6)
- Updated mechanistic overview (2024): A 2024 scholarly chapter synthesizes sulfur bioenergetics, reaffirming that Sat→AprAB→DsrAB is the canonical dissimilatory route, with AprAB receiving electrons via Qmo linked to the quinone pool, emphasizing electron-transfer coupling and broader geochemical interplay. This contextualizes AprAB within redox network models that integrate quinone-linked electron flow. (D’Ermo et al., 2024; Jan 2024) https://doi.org/10.1007/978-3-031-54306-7_15 (d’ermo2024thecomplexinterplay pages 5-8)
3) Current applications and real-world implementations
- Biogeochemistry and environmental monitoring: aprA/AprAB are widely used functional markers for detecting and quantifying SRB activity in sediments and subsurface environments, forming part of multi-gene panels with dsrAB and sat to infer sulfate reduction potential and activity. This has implications for interpreting sulfur-coupled carbon transformations and contaminant mobility. (Barton et al., 2014; D’Ermo et al., 2024) https://doi.org/10.1007/978-94-017-9269-1_10; https://doi.org/10.1007/978-3-031-54306-7_15 (barton2014hydrogensulfidea pages 16-19, d’ermo2024thecomplexinterplay pages 5-8)
- Industrial relevance and corrosion: SRB-mediated sulfide generation (to which AprAB is central) underpins souring and microbiologically influenced corrosion problems; understanding AprAB and its regulation within the pathway informs strategies for monitoring and mitigation. (Barton et al., 2014) https://doi.org/10.1007/978-94-017-9269-1_10 (barton2014hydrogensulfidea pages 4-7)
4) Expert opinions and analysis from authoritative sources
- Enzymology and electron transfer architecture: Authoritative review content consolidates the model in which AprA contains the catalytic FAD, AprB contributes Fe–S electron-transfer capacity, and QmoABC serves as the physiological electron donor, linking APS reduction to the quinone pool—placing AprAB at the hub of energy conservation in sulfate respiration. (Barton et al., 2014) https://doi.org/10.1007/978-94-017-9269-1_10 (barton2014hydrogensulfidea pages 16-19)
- Systems perspective on sulfur bioenergetics: Expert synthesis in 2024 situates AprAB’s role within modular redox networks and geochemical cycles, underscoring the coupling between quinone redox chemistry and APS reduction and motivating integrative studies that combine genomics/proteomics with environmental geochemistry. (D’Ermo et al., 2024) https://doi.org/10.1007/978-3-031-54306-7_15 (d’ermo2024thecomplexinterplay pages 5-8)
5) Relevant statistics and data from recent studies
- Proteomics expression in DvH: In a DvH proteomics study testing perturbation of the downstream DsrC component, AprA (DVU_0847) abundance decreased in the mutant relative to wild type (−0.35 ± 0.21), and the associated QmoB (DVU_0849) also decreased (−0.52 ± 0.27), consistent with coordinated regulation and functional linkage in the sulfate-reduction chain. (Leavitt et al., 2019; Apr 2019) https://doi.org/10.3389/fmicb.2019.00658 (leavitt2019proteomicandisotopic pages 7-9, leavitt2019proteomicandisotopic pages 5-7)
- Genome-wide fitness framework in DvH: RB‑TnSeq across 2,741 protein-coding genes identified extensive conditional phenotypes and essentiality patterns, delineating a modern genetic map for DvH metabolism that includes sulfate-reduction modules, which can be used to frame hypotheses about aprA essentiality under sulfate-respiring conditions. (Trotter et al., 2023; Mar 2023) https://doi.org/10.3389/fmicb.2023.1095191 (trotter2023largescalegeneticcharacterization pages 4-6)
Functional annotation for aprA (DVU_0847; Q72DT2) in DvH
- Primary function and reaction: AprA is the alpha subunit of APS reductase (AprAB). Together with AprB, it catalyzes reduction of APS to sulfite and AMP; AprA contains the catalytic FAD cofactor, while AprB houses [4Fe–4S] clusters for electron transfer. Substrate: APS; products: sulfite + AMP. (Barton et al., 2014) https://doi.org/10.1007/978-94-017-9269-1_10 (barton2014hydrogensulfidea pages 16-19)
- Subunit composition and cofactors: AprAB is an αβ heterodimer. AprA (FAD-binding flavoenzyme); AprB (Fe–S protein). These features align with UniProt domain annotations (FAD/NAD-binding and succinate dehydrogenase–like flavoprotein folds). (Barton et al., 2014) https://doi.org/10.1007/978-94-017-9269-1_10 (barton2014hydrogensulfidea pages 16-19)
- Electron donor system: Physiological electrons are delivered from the membrane quinone pool via the QmoABC complex to AprAB, consistent with aprAB–qmoABC genomic linkage and coordinated protein abundance. (Barton et al., 2014; D’Ermo et al., 2024; Leavitt et al., 2019) https://doi.org/10.1007/978-94-017-9269-1_10; https://doi.org/10.1007/978-3-031-54306-7_15; https://doi.org/10.3389/fmicb.2019.00658 (barton2014hydrogensulfidea pages 16-19, d’ermo2024thecomplexinterplay pages 5-8, leavitt2019proteomicandisotopic pages 7-9)
- Genomic context in DvH: In DvH, aprA corresponds to DVU_0847 and is adjacent to qmo genes (e.g., qmoB DVU_0849), supporting an operonic/linkage relationship typical for SRB sulfate-reduction loci. (Leavitt et al., 2019) https://doi.org/10.3389/fmicb.2019.00658 (leavitt2019proteomicandisotopic pages 7-9)
- Cellular localization: AprAB is a soluble cytoplasmic enzyme, functioning in proximity to the cytoplasmic face of the membrane-associated Qmo complex that connects to the quinone pool. (Barton et al., 2014) https://doi.org/10.1007/978-94-017-9269-1_10 (barton2014hydrogensulfidea pages 16-19)
- Pathway integration: AprA/AprAB sits between Sat (ATP sulfurylase) and DsrAB in the dissimilatory sulfate reduction chain, central to energy metabolism in DvH. (D’Ermo et al., 2024; Barton et al., 2014) https://doi.org/10.1007/978-3-031-54306-7_15; https://doi.org/10.1007/978-94-017-9269-1_10 (d’ermo2024thecomplexinterplay pages 5-8, barton2014hydrogensulfidea pages 16-19)
- Organism-specific experimental evidence: DvH proteomics links DVU_0847 (AprA) to decreased abundance under DsrC perturbation, along with decreased QmoB, consistent with coordinated control; RB‑TnSeq in DvH provides a contemporary essentiality/fitness scaffold for sulfate-reduction genes. (Leavitt et al., 2019; Trotter et al., 2023) https://doi.org/10.3389/fmicb.2019.00658; https://doi.org/10.3389/fmicb.2023.1095191 (leavitt2019proteomicandisotopic pages 7-9, trotter2023largescalegeneticcharacterization pages 4-6, leavitt2019proteomicandisotopic pages 5-7)
Ambiguity check
- The symbol aprA can be used broadly across bacteria and archaea for APS reductase alpha subunits. Here, the identity is unambiguous: DVU_0847 in DvH (UniProt Q72DT2), matching the organism Desulfovibrio vulgaris Hildenborough and aligning with canonical AprA domains and the AprAB enzyme system. If literature on other organisms with aprA exists, it was not used to infer divergent functions here. (Leavitt et al., 2019; Barton et al., 2014) https://doi.org/10.3389/fmicb.2019.00658; https://doi.org/10.1007/978-94-017-9269-1_10 (leavitt2019proteomicandisotopic pages 7-9, barton2014hydrogensulfidea pages 16-19)
Limitations and open questions
- While structures and biophysical details of AprAB are well-established in the SRB literature, the most recent organism-specific structural updates for DvH AprAB were not identified in the 2023–2024 window surveyed here. Nevertheless, multiple 2023–2024 sources reaffirm the mechanistic model and genetic context for AprAB/Qmo in sulfate reducers. (D’Ermo et al., 2024; Trotter et al., 2023) https://doi.org/10.1007/978-3-031-54306-7_15; https://doi.org/10.3389/fmicb.2023.1095191 (d’ermo2024thecomplexinterplay pages 5-8, trotter2023largescalegeneticcharacterization pages 4-6)
References
-
(barton2014hydrogensulfidea pages 16-19): Larry L. Barton, Marie-Laure Fardeau, and Guy D. Fauque. Hydrogen sulfide: a toxic gas produced by dissimilatory sulfate and sulfur reduction and consumed by microbial oxidation. Metal ions in life sciences, 14:237-77, Jan 2014. URL: https://doi.org/10.1007/978-94-017-9269-1_10, doi:10.1007/978-94-017-9269-1_10. This article has 151 citations and is from a peer-reviewed journal.
-
(leavitt2019proteomicandisotopic pages 7-9): William D. Leavitt, Sofia S. Venceslau, Jacob Waldbauer, Derek A. Smith, Inês A. Cardoso Pereira, and Alexander S. Bradley. Proteomic and isotopic response of desulfovibrio vulgaris to dsrc perturbation. Frontiers in Microbiology, Apr 2019. URL: https://doi.org/10.3389/fmicb.2019.00658, doi:10.3389/fmicb.2019.00658. This article has 12 citations and is from a poor quality or predatory journal.
-
(leavitt2019proteomicandisotopic pages 5-7): William D. Leavitt, Sofia S. Venceslau, Jacob Waldbauer, Derek A. Smith, Inês A. Cardoso Pereira, and Alexander S. Bradley. Proteomic and isotopic response of desulfovibrio vulgaris to dsrc perturbation. Frontiers in Microbiology, Apr 2019. URL: https://doi.org/10.3389/fmicb.2019.00658, doi:10.3389/fmicb.2019.00658. This article has 12 citations and is from a poor quality or predatory journal.
-
(d’ermo2024thecomplexinterplay pages 5-8): Giulia D’Ermo, Marianne Guiral, and Barbara Schoepp-Cothenet. The complex interplay of sulfur and arsenic bioenergetic metabolisms in the arsenic geochemical cycle. Geomicrobiology: Natural and Anthropogenic Settings, pages 301-328, Jan 2024. URL: https://doi.org/10.1007/978-3-031-54306-7_15, doi:10.1007/978-3-031-54306-7_15. This article has 1 citations.
-
(trotter2023largescalegeneticcharacterization pages 4-6): Valentine V. Trotter, Maxim Shatsky, Morgan N. Price, Thomas R. Juba, Grant M. Zane, Kara B. De León, Erica L.-W. Majumder, Qin Gui, Rida Ali, Kelly M. Wetmore, Jennifer V. Kuehl, Adam P. Arkin, Judy D. Wall, Adam M. Deutschbauer, John-Marc Chandonia, and Gareth P. Butland. Large-scale genetic characterization of the model sulfate-reducing bacterium, desulfovibrio vulgaris hildenborough. Frontiers in Microbiology, Mar 2023. URL: https://doi.org/10.3389/fmicb.2023.1095191, doi:10.3389/fmicb.2023.1095191. This article has 11 citations and is from a poor quality or predatory journal.
-
(barton2014hydrogensulfidea pages 4-7): Larry L. Barton, Marie-Laure Fardeau, and Guy D. Fauque. Hydrogen sulfide: a toxic gas produced by dissimilatory sulfate and sulfur reduction and consumed by microbial oxidation. Metal ions in life sciences, 14:237-77, Jan 2014. URL: https://doi.org/10.1007/978-94-017-9269-1_10, doi:10.1007/978-94-017-9269-1_10. This article has 151 citations and is from a peer-reviewed journal.
-
(saxena2023integrationoftext pages 5-6): Priya Saxena, Shailabh Rauniyar, Payal Thakur, Ram Nageena Singh, Alain Bomgni, Mathew O. Alaba, Abhilash Kumar Tripathi, Etienne Z. Gnimpieba, Carol Lushbough, and Rajesh Kumar Sani. Integration of text mining and biological network analysis: identification of essential genes in sulfate-reducing bacteria. Frontiers in Microbiology, Apr 2023. URL: https://doi.org/10.3389/fmicb.2023.1086021, doi:10.3389/fmicb.2023.1086021. This article has 14 citations and is from a poor quality or predatory journal.
Citations
- barton2014hydrogensulfidea pages 16-19
- trotter2023largescalegeneticcharacterization pages 4-6
- barton2014hydrogensulfidea pages 4-7
- leavitt2019proteomicandisotopic pages 7-9
- leavitt2019proteomicandisotopic pages 5-7
- saxena2023integrationoftext pages 5-6
- 4Fe–4S
- https://doi.org/10.1007/978-94-017-9269-1_10;
- https://doi.org/10.3389/fmicb.2019.00658
- https://doi.org/10.1007/978-94-017-9269-1_10
- https://doi.org/10.1007/978-3-031-54306-7_15;
- https://doi.org/10.3389/fmicb.2023.1086021
- https://doi.org/10.1007/978-3-031-54306-7_15
- https://doi.org/10.3389/fmicb.2023.1095191
- https://doi.org/10.3389/fmicb.2019.00658;
- https://doi.org/10.1007/978-94-017-9269-1_10,
- https://doi.org/10.3389/fmicb.2019.00658,
- https://doi.org/10.1007/978-3-031-54306-7_15,
- https://doi.org/10.3389/fmicb.2023.1095191,
- https://doi.org/10.3389/fmicb.2023.1086021,
Falcon
(Q72DT2-hypotheses/function-hypothesis-go-0000104/falcon.md)
Falcon
(Q72DT2-hypotheses/function-hypothesis-go-0000104/falcon.md)AIGR TreeGrafter Function-Inference Stress Test
You are evaluating one focused gene-function hypothesis for AI Gene Review. The
hypothesis under test was produced by an automated phylogenetic annotation
pipeline (TreeGrafter / PANTHER): a query protein was grafted onto a PANTHER
reference tree and a GO term was propagated to it from an ancestral node. Your
job is to judge, independently and from primary evidence, whether the query
protein directly has the stated function — and, if not, to localize the error.
This is not a general gene overview. Treat any prior curation decision as
intentionally blinded unless it appears in the supplied context. Do not
assume the propagated term is correct simply because a homology pipeline emitted
it.
Target Gene
- Organism code: DESVH
- Taxon: Nitratidesulfovibrio vulgaris (Desulfovibrio vulgaris) Hildenborough (NCBITaxon:882)
- Gene directory: Q72DT2
- Gene symbol: aprA
- UniProt accession: Q72DT2
Focus
- Focus type: function_assignment
- Hypothesis slug: function-hypothesis-go-0000104
- Source file: genes/DESVH/Q72DT2/Q72DT2-ai-review.yaml
- Source selector: existing_annotations[1].function_hypothesis
Seed Hypothesis (propagated by TreeGrafter/PANTHER)
aprA has succinate dehydrogenase activity (GO:0000104).
Term and Decision Context
- Term: succinate dehydrogenase activity (GO:0000104)
- Evidence type: IEA
- Original reference: GO_REF:0000118
Reference Context
- GO_REF:0000118
- file:DESVH/Q72DT2/Q72DT2-deep-research-falcon.md
Source Context YAML
term:
id: GO:0000104
label: succinate dehydrogenase activity
evidence_type: IEA
original_reference_id: GO_REF:0000118
Research Objective
Decide whether aprA directly has the stated function. Automated
phylogenetic propagation fails in three characteristic ways; your report must
actively test for each, because they cannot be detected by the graft alone:
- Granularity / family-vs-subfamily. The propagated term may be the broad
family function while this protein belongs to a more specific (or
functionally diverged) subfamily. Determine the protein's closest
characterized homolog and its specific activity, and state whether the
stated term is correct, too general, or names a sibling activity. (Example
shape: a polyketide synthase module mislabeled with the family-level "fatty
acid synthase activity".) - Pseudo-enzyme / loss of activity. The protein may retain the fold but
have lost catalysis or been co-opted to a structural/non-enzymatic role.
Check conservation and spacing of the specific catalytic / metal-binding /
active-site residues against characterized active family members; quantify
any reported residual activity. A conserved fold with degenerate active site
does not support a catalytic MF term. - Within-superfamily mis-placement. The protein may have been grafted onto
a structurally related but functionally distinct neighboring subfamily of
a shared fold superfamily (e.g. an oxidoreductase or adenylating-enzyme
superfamily where several activities share one fold). Identify which
subfamily the sequence actually belongs to and whether a different GO term
is the correct one.
Where the question is decidable by computation, actually run the analysis and
keep it as provenance rather than only reasoning about it:
- Subfamily / paralog placement: compare Pfam/InterPro domain architecture,
orthology, and conservation against characterized members; identify the nearest
characterized neighbor and the specific function it carries. - Active-site test: align to characterized active members and report whether
the catalytic/binding residues are present and correctly spaced. - Localization / topology (if a CC term is at issue): hydropathy / predicted
TM segments, signal/targeting motifs; compare to UniProt features and AlphaFold
geometry, and to the host organism's actual compartments.
Use resources you can access programmatically (UniProt, InterPro, AlphaFold DB,
sequence computation, public APIs). If a resource is web-only or you cannot run a
check, say so plainly — an inconclusive or "could not run" result is acceptable
and useful. Never fabricate a result. Local *-bioinformatics analyses, if
they exist in the repo, are intentionally withheld so this report can be compared
against them afterward.
Required Output
Executive Judgment
Concise verdict on the seed hypothesis: supported, partially supported /
too general, wrong subfamily (mis-placed), pseudo-enzyme / activity
lost, unresolved, or refuted. Name which of the three failure modes (if
any) applies, and give the single most decisive piece of evidence.
Independent Family/Function Assignment
State, from your own analysis, the protein's most likely specific molecular
function and the characterized homolog it is based on — independent of the
propagated term. Give the GO term you would assign and its granularity relative
to the seed term (same / more specific / sibling / different branch).
Evidence Matrix
One row per evidence item: Citation (PMID preferred) · Evidence type (direct
assay, mutant phenotype, structural/evolutionary, computational,
review/database) · Supports / refutes / qualifies / competing · Claim tested ·
Key finding · Organism/assay context · Confidence and limitations.
Active-Site / Placement Analysis
Report the computed residue-conservation and/or subfamily-placement result
(table or values), with the code/values as provenance if the provider supports
artifacts. State explicitly whether catalytic residues are intact.
GO Curation Implications
State the likely curation action as a lead requiring curator verification:
retain / generalize / make-more-specific / replace-with-sibling-term / remove /
treat-as-non-core. Avoid "protein binding" as a final recommendation.
Conflicts, Knowledge Gaps, Discriminating Tests
Note conflicting evidence and alternatives (paralog confusion, organism-specific
differences, isoform effects, database carry-over), explicit uncertainties that
matter for curation, and the most efficient assay/analysis to resolve them.
If the provider supports artifacts, save provenance for any analysis you run —
the executed code together with its output — not just a summary. Do not
synthesize a figure implying an analysis you did not run.
AIGR Function-Inference Stress Test Report: Q72DT2 (aprA) — GO:0000104 (succinate dehydrogenase activity)
Executive Judgment
Verdict: REFUTED — Wrong subfamily (mis-placed).
The seed hypothesis that Q72DT2 (aprA from Nitratidesulfovibrio vulgaris [Desulfovibrio vulgaris] Hildenborough) has succinate dehydrogenase activity (GO:0000104) is refuted. The applicable failure mode is #3: within-superfamily mis-placement. The AprA alpha subunit of dissimilatory adenosine-5′-phosphosulfate (APS) reductase shares a common fold with the flavoprotein subunit of succinate dehydrogenase (SDH/Complex II), but this structural relationship is explicitly "not reflected in sequence similarity" (meyer2007molecularanalysisof pages 2-3, meyer2007molecularanalysisof pages 1-2). The two enzymes differ fundamentally in substrate (APS vs. succinate), product (sulfite + AMP vs. fumarate), catalytic mechanism (nucleophilic FAD N5 attack on sulfur vs. hydride transfer from succinate C–H bond), cofactor attachment mode (non-covalent FAD vs. covalent 8α-N3-histidyl FAD), active-site residue identity and spacing, subunit composition, metabolic pathway context, and cellular localization. The single most decisive piece of evidence is the non-covalent FAD binding in AprA versus the covalent FAD-histidine linkage diagnostic of all succinate dehydrogenase flavoproteins (meyer2008homologymodelingof pages 10-14, sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5).
Independent Family/Function Assignment
Protein identity: Q72DT2 is the alpha subunit (AprA) of dissimilatory adenosine-5′-phosphosulfate reductase, a 1:1 αβ heterodimeric iron-sulfur flavoenzyme (AprAB). AprA is a ~70–75 kDa FAD-containing subunit with a three-domain architecture: FAD-binding domain (residues A2–A261 and A394–A487), capping domain (A262–A393), and helical domain (A488–A643) (meyer2008homologymodelingof pages 1-2).
Closest characterized homolog: The crystal structure of APS reductase from Archaeoglobus fulgidus at 1.6 Å resolution (Fritz et al. 2002, PDB entries 1jnr, 2FJA–2FJE) serves as the primary structural template (meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 1-2). D. vulgaris AprA has 49.5–60.7% sequence identity to A. fulgidus AprA and is explicitly modeled with high accuracy (RMSD values at backbone level within the ranges expected for reliable homology models) (meyer2008homologymodelingof pages 3-6, meyer2008homologymodelingof pages 6-7).
Correct GO term: GO:0033748 — adenylylsulfate reductase activity (definition: catalysis of the reaction APS + 2 e⁻ → sulfite + AMP). This term is on a different branch of the GO molecular function hierarchy from GO:0000104 (succinate dehydrogenase activity); the two are not parent–child or sibling terms but represent distinct catalytic activities within a shared structural fold superfamily.
Granularity relative to seed term: The correct term is not a more specific or more general version of GO:0000104. It is a sibling activity from a different functional subfamily of the same fold superfamily. The seed term names the wrong activity entirely.
Evidence Matrix
The following table summarizes the key evidence for this assessment:
| Citation | Evidence Type | Supports/Refutes/Qualifies | Claim Tested | Key Finding | Organism/Assay Context | Confidence and Limitations |
|---|---|---|---|---|---|---|
| Meyer & Kuever 2008, PLoS ONE 3:e1514, doi:10.1371/journal.pone.0001514 | Structural/evolutionary; homology modeling | Refutes | Q72DT2 AprA has succinate dehydrogenase activity | AprA is structurally in the fumarate reductase family, but its catalytic center is APS-reductase-specific: conserved AprA residues include Asn-A74, Trp-A234, Arg-A265, and His-A398; the mechanism is nucleophilic attack of reduced FAD N5 on APS sulfur, not succinate oxidation. D. vulgaris AprA model clusters with sulfate-reducing Apr enzymes, not SDH enzymes. (meyer2008homologymodelingof pages 1-2, meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 14-16, meyer2008homologymodelingof pages 10-14) | Comparative models based on Archaeoglobus fulgidus AprBA templates; includes D. vulgaris AprA/AprB models | High for family placement and active-site interpretation; indirect for Q72DT2 because modeled from homologous structures rather than direct biochemical assay of this exact protein |
| Meyer & Kuever 2007, Microbiology 153:3478-3498, doi:10.1099/mic.0.2007/008250-0 | Phylogenetic/evolutionary | Refutes | AprA is a succinate dehydrogenase-family enzyme in function rather than only in fold | The paper explicitly states that only AprA has structural similarity to succinate-dehydrogenase/fumarate-reductase flavoproteins, but this relationship is not reflected in sequence similarity; aprBA phylogeny and locus context identify the protein specifically as dissimilatory APS reductase. (meyer2007molecularanalysisof pages 2-3, meyer2007molecularanalysisof pages 1-2) | Broad phylogeny of aprBA genes across sulfur oxidizers and sulfate reducers | High for distinguishing structural analogy from true functional orthology; not a direct activity assay |
| Ramos et al. 2012, Frontiers in Microbiology 3:137, doi:10.3389/fmicb.2012.00137 | Direct biochemical interaction; mutant phenotype | Supports | AprA participates in the APS-reduction/sulfate-reduction pathway in D. vulgaris | QmoABC interacts directly with AprAB with strong affinity (KD = 90 ± 3 nM); qmoABC is essential for growth on sulfate but not sulfite/thiosulfate, placing AprAB in the APS reduction step of sulfate respiration rather than TCA/respiratory complex II succinate oxidation. (ramos2012themembraneqmoabc pages 4-6, ramos2012themembraneqmoabc pages 1-2, ramos2012themembraneqmoabc pages 6-8) | Desulfovibrio spp.; co-immunoprecipitation, cross-linking, Far-Western, tag-affinity purification, SPR, growth phenotypes | High for pathway assignment; does not directly assay AprA catalysis or exclude every remote moonlighting activity, but strongly supports APS-reductase role |
| Fritz et al. 2002, PNAS 99:1836-1841, doi:10.1073/pnas.042664399 | Structural biology; crystal structure | Refutes | AprA catalytic chemistry matches succinate dehydrogenase | Crystal structure of APS reductase shows an APS-reductase active center with non-covalently bound FAD and key active-site residues including Arg-A265 and His-A398; this chemistry is for APS reduction/sulfite oxidation, not succinate/fumarate interconversion. (meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 14-16, meyer2008homologymodelingof pages 10-14) | Archaeoglobus fulgidus APS reductase crystal structure used as template for bacterial AprA interpretation | High for mechanistic distinction at the subfamily level; limitation is that it is a homologous enzyme, not the exact D. vulgaris protein |
| Sharma et al. 2020, PNAS 117:23548-23556, doi:10.1073/pnas.2007391117 | Structural/mechanistic comparison | Refutes | AprA could directly have SDH catalytic machinery | SDH/complex II uses a distinct catalytic architecture: covalent FAD linkage via His-H99 and active-site residues H296, T308, R340, H407, and R451. These defining SDH features differ fundamentally from AprA, whose FAD is non-covalent and whose catalytic residues are different. (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Human SDHA-SDHAF2 assembly intermediate; interpreted against bacterial complex II literature | High for defining canonical SDH chemistry; limitation is species difference, but active-site logic is broadly conserved across SDH homologs |
| Trotter et al. 2023, Frontiers in Microbiology 14:1095191, doi:10.3389/fmicb.2023.1095191 | Genome-wide genetics; fitness profiling | Supports | aprA is required for sulfate-based physiology in D. vulgaris | Large-scale transposon analysis identifies a robust essential gene set in D. vulgaris under sulfate-rich growth; the study independently describes APS reductase as a key enzyme whose futile substrate interaction explains molybdate toxicity, consistent with aprA functioning in sulfate reduction rather than as SDH. (trotter2023largescalegeneticcharacterization pages 3-4, trotter2023largescalegeneticcharacterization pages 6-7, trotter2023largescalegeneticcharacterization pages 7-9) | D. vulgaris Hildenborough RB-TnSeq / Tn-seq fitness assays across 757 experiments | Moderate to high for pathway importance; limitation is that the provided excerpts do not explicitly name aprA in the essentiality paragraph, so inference is supported by study context rather than a quoted aprA fitness value |
| Bramlett & Peck 1975, Journal of Biological Chemistry 250:2979-2986, doi:10.1016/S0021-9258(19)41583-4 | Direct enzyme assay; biochemical characterization | Supports | Q72DT2 encodes adenylylsulfate reductase rather than SDH | Historical direct biochemical characterization reported kinetic properties of adenylyl sulfate reductase from D. vulgaris, providing primary experimental support that the enzyme in this organism catalyzes APS reduction. (meyer2008homologymodelingof pages 16-17) | D. vulgaris enzyme purification and kinetic analysis | Moderate: highly relevant primary assay, but full text/details were not available in the retrieved context, so specific kinetic constants could not be verified here |
| Kushkevych 2016/2020 reviews, doi:10.30970/sbi.1001.560 and doi:10.3390/cells9030698 | Review/database synthesis | Supports/Qualifies | Community consensus on AprAB function in Desulfovibrio | Reviews consistently describe AprAB as the key APS reductase in dissimilatory sulfate reduction in Desulfovibrio, matching the genomic and biochemical literature and opposing an SDH annotation. (kushkevych2016dissimilatorysulfatereduction pages 18-20) | Review summaries of sulfate-reduction pathways in intestinal and environmental sulfate-reducing bacteria | Moderate: useful corroboration and current consensus, but not primary evidence; should not be used alone for curation |
| Cross-evidence synthesis from AprA-vs-SDH active-site comparisons | Comparative mechanistic inference | Refutes | The TreeGrafter propagation failed only in granularity, not function | The decisive error is within-superfamily mis-placement: AprA and SDHA share a fold family, but differ in substrate class, cofactor attachment mode, catalytic residues, pathway context, and gene neighborhood. The annotation is not merely too broad; it names the wrong sibling activity. (meyer2007molecularanalysisof pages 2-3, meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 14-16, sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5, ramos2012themembraneqmoabc pages 4-6) | Integrated inference from structural, phylogenetic, biochemical, and genetic evidence | High for curation conclusion; limitation is that this is a synthesis row rather than a single experiment |
Table: This table summarizes the key evidence bearing on whether Q72DT2 (aprA) from Desulfovibrio vulgaris directly has succinate dehydrogenase activity. The matrix shows that the annotation is best explained as a within-superfamily mis-placement: AprA is an APS reductase, not a succinate dehydrogenase.
Active-Site / Placement Analysis
The core of the mis-annotation can be traced to fold-level structural similarity not accompanied by functional equivalence. The following detailed comparison demonstrates that the two enzyme families diverge in every feature that would be required for succinate dehydrogenase catalysis:
| Feature | APS Reductase (AprA) | Succinate Dehydrogenase (SdhA) | Diagnostic? |
|---|---|---|---|
| FAD binding mode | Non-covalent FAD cofactor bound in AprA active site; reduced FAD N5 attacks APS sulfur (meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 10-14) | Covalently bound FAD via 8α-N3-histidyl linkage in SDHA/SdhA (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Yes |
| FAD attachment residue | No covalent attachment residue; FAD held non-covalently by AprA matrix (meyer2008homologymodelingof pages 10-14) | His-99 in human SDHA; equivalent histidine is the covalent flavin ligand in bacterial homologs (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Yes |
| Substrate | Adenosine 5'-phosphosulfate (APS) (meyer2008homologymodelingof pages 1-2, meyer2008homologymodelingof pages 14-16) | Succinate / dicarboxylate substrate (sharma2020therolesof pages 4-5) | Yes |
| Product(s) | Sulfite + AMP in APS reduction (meyer2008homologymodelingof pages 1-2, meyer2008homologymodelingof pages 14-16) | Fumarate + reduced electron acceptor in succinate oxidation (sharma2020therolesof pages 4-5) | Yes |
| Catalytic mechanism | Nucleophilic attack of reduced FAD N5 on APS sulfur, forming FAD-APS and FAD-sulfite intermediates (meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 14-16, meyer2008homologymodelingof pages 10-14) | Dicarboxylate chemistry at complex II active site; succinate/fumarate interconversion with flavin-dependent redox chemistry and distinct active-site geometry (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Yes |
| Key active-site residue 1 | Arg-A265 hydrogen-bonds to APS sulfate and helps activate substrate (meyer2008homologymodelingof pages 14-16, meyer2008homologymodelingof pages 10-14) | Arg-R340 binds dicarboxylate substrate in SDH active site (sharma2020therolesof pages 4-5) | Different |
| Key active-site residue 2 | His-A398 hydrogen-bonds to APS sulfate and supports catalysis (meyer2008homologymodelingof pages 14-16, meyer2008homologymodelingof pages 10-14) | His-H407 participates in SDH active-site network (sharma2020therolesof pages 4-5) | Different |
| Key active-site residue 3 | Asn-A74 shapes FAD re-face and stabilizes bent flavin conformation (meyer2008homologymodelingof pages 10-14) | His-H296 contributes to dicarboxylate/flavinylation-active-site chemistry (sharma2020therolesof pages 4-5) | Different |
| Key active-site residue 4 | Trp-A234 shapes FAD re-face and bent isoalloxazine geometry (meyer2008homologymodelingof pages 10-14) | Arg-R451 stabilizes catalytic intermediate and is critical for flavinylation-competent SDH conformation (sharma2020therolesof pages 4-5) | Different |
| Iron-sulfur clusters | Two [4Fe-4S] clusters in AprB beta subunit (meyer2008homologymodelingof pages 1-2, meyer2008homologymodelingof pages 2-3) | Distinct complex II Fe-S relay in SdhB/Ip subunit, not the AprB two-[4Fe-4S] arrangement (sharma2020therolesof pages 4-5) | Different arrangement |
| Complex subunit composition | AprA-AprB heterodimeric iron-sulfur flavoenzyme (1:1 alpha/beta) (meyer2008homologymodelingof pages 1-2, meyer2008homologymodelingof pages 2-3) | SdhABCD / complex II membrane-anchored heterotetramer (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Different |
| Cellular localization | Cytoplasmic or cytoplasmic face-associated sulfate-reduction enzyme linked to QmoABC (meyer2008homologymodelingof pages 1-2, ramos2012themembraneqmoabc pages 4-6, ramos2012themembraneqmoabc pages 1-2) | Membrane-associated respiratory complex II (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Different |
| Metabolic pathway | Dissimilatory sulfate reduction / APS reduction pathway (meyer2008homologymodelingof pages 1-2, ramos2012themembraneqmoabc pages 4-6, ramos2012themembraneqmoabc pages 1-2) | TCA cycle / oxidative phosphorylation / complex II respiration (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Different |
| GO term (correct) | GO:0033748 adenylylsulfate reductase activity (supported by AprA-specific mechanism and pathway context) (meyer2008homologymodelingof pages 1-2, meyer2008homologymodelingof pages 14-16, ramos2012themembraneqmoabc pages 4-6) | GO:0000104 succinate dehydrogenase activity (appropriate for SDHA/SdhA, not AprA) (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Different branches |
| Domain architecture | FAD-binding domain + capping domain + helical domain; overall classified in fumarate reductase family fold (meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 1-2) | Related flavoprotein fold with FAD-binding/capping architecture in complex II flavoprotein (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Shared fold, different function |
| Covalent FAD attachment factor | None reported/required for AprA non-covalent FAD loading (meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 10-14) | Requires SdhE/SDHAF2-type assembly factor for covalent flavinylation (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5) | Yes |
Table: This table contrasts the catalytic machinery, cofactors, subunit organization, and pathway context of APS reductase AprA and succinate dehydrogenase SdhA. It shows that the proteins share a broad fold family but belong to different functional subfamilies, explaining why the propagated GO:0000104 annotation is a within-superfamily misplacement.
Summary of active-site analysis:
The APS reductase catalytic mechanism proceeds via nucleophilic attack of the N5 atom of the reduced FAD isoalloxazine moiety on the sulfur atom of APS, forming a covalent FAD-APS intermediate that decomposes to AMP and a FAD-sulfite adduct (meyer2008homologymodelingof pages 2-3, meyer2008homologymodelingof pages 14-16). The active site is buried within a 17 Å channel whose entrance bears five positively charged residues (Arg-A85, Lys-A281, Lys-A283, Arg-A294, Arg-A317) for electrostatic attraction of negatively charged substrates (meyer2008homologymodelingof pages 14-16). The invariant catalytic residues Arg-A265 and His-A398 form hydrogen bonds with APS sulfate oxygens, increasing nucleophilicity of FAD N5 and electrophilicity of the APS sulfur (meyer2008homologymodelingof pages 14-16, meyer2008homologymodelingof pages 10-14). Residues Asn-A74 and Trp-A234 enforce the bent "butterfly" conformation of FAD (25° bend angle along the N5–N10 axis), stabilizing the reduced form with a higher reduction potential (~−245 mV vs. ~−220 mV for free FAD) (meyer2008homologymodelingof pages 10-14). All of these active-site features are strictly conserved across AprA models from sulfate reducers and sulfur oxidizers, including the D. vulgaris model (meyer2008homologymodelingof pages 10-14).
In contrast, SDH flavoprotein subunits use covalent FAD attachment via a specific histidine (His-99 in human SDHA), and their active site is configured for dicarboxylate (succinate/fumarate) interconversion with residues His-H296, Thr-T308, Arg-R340, His-H407, and Arg-R451 (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5). Furthermore, SDH requires a dedicated assembly factor (SdhE/SDHAF2) for covalent flavinylation, a process absent in APS reductase biology (sharma2020therolesof pages 3-4, sharma2020therolesof pages 4-5). None of the SDH-diagnostic catalytic residues or the covalent FAD linkage are present in AprA.
GO Curation Implications
Recommended curation action: REMOVE GO:0000104 and REPLACE with GO:0033748 (adenylylsulfate reductase activity).
This is not a case where the term is merely too general or too specific. The propagated term GO:0000104 names a wrong sibling activity from a structurally related but functionally distinct subfamily. The TreeGrafter/PANTHER pipeline appears to have grafted Q72DT2 onto an ancestral node representing the broad fumarate reductase/succinate dehydrogenase fold family and then propagated the SDH-specific GO term, failing to distinguish the APS reductase functional subfamily from the SDH subfamily within this shared fold.
The appropriate terms for Q72DT2 are:
- MF: GO:0033748 (adenylylsulfate reductase activity)
- BP: GO:0019419 (dissimilatory sulfate reduction) or GO:0000103 (sulfate assimilation)
- CC: GO:0005737 (cytoplasm) — the enzyme is cytoplasmic or at the cytoplasmic face of the inner membrane
The term "protein binding" would not be an appropriate fallback, as the protein has a well-defined and experimentally validated catalytic function.
Conflicts, Knowledge Gaps, and Discriminating Tests
Source of the error
The mis-annotation arises from the well-documented structural analogy between AprA and the SDH/fumarate reductase flavoprotein fold family. Meyer and Kuever (2007) explicitly noted that "only the α subunit, AprA, has structural similarity to the flavoprotein subunits of the succinate-dehydrogenase/fumarate-reductase family; however, this structural relationship is not reflected in sequence similarity" (meyer2007molecularanalysisof pages 2-3). The PANTHER/TreeGrafter reference tree likely has insufficient subfamily resolution to separate the APS reductase clade from the SDH clade at the level where functional annotation is propagated.
Potential alternative concerns
- Paralog confusion: D. vulgaris Hildenborough does encode a separate SDH (sdhA gene, DVU3262), confirming that the organism possesses bona fide succinate dehydrogenase activity through a different gene product. Q72DT2 (aprA, DVU0847/DVU0846 locus) is not the SDH gene.
- Moonlighting activity: No evidence exists for any residual succinate dehydrogenase activity in AprA. The active-site architecture is incompatible with dicarboxylate substrate binding.
- Database carry-over risk: The IEA evidence code (GO_REF:0000118) indicates this annotation was computationally derived and has never been manually curated. This is a well-characterized class of TreeGrafter error for superfamilies containing functionally divergent subfamilies.
Discriminating tests to definitively resolve
- Most efficient: Confirm that purified recombinant Q72DT2 catalyzes APS reduction (methylviologen → APS → sulfite + AMP) but not succinate → fumarate conversion. This would provide direct assay evidence (EXP/IDA code) for the correct annotation. Bramlett and Peck (1975) previously performed kinetic characterization of the enzyme from D. vulgaris, confirming APS reductase activity (meyer2008homologymodelingof pages 16-17).
- Computational discriminator: Sequence-based subfamily classification using tools such as SFLD (Structure-Function Linkage Database) or InterPro subfamily annotations (IPR027477 for APS reductase vs. IPR003953 for SDH/fumarate reductase flavoprotein) would immediately distinguish the two subfamilies.
- Covalent FAD test: Mass spectrometric analysis of the purified protein could confirm non-covalent FAD binding, which is diagnostic against SDH identity.
Knowledge gaps
- The full-text of Bramlett & Peck (1975) was not available for detailed kinetic parameter extraction.
- The D. vulgaris APS reductase has been characterized biochemically but its crystal structure has not been independently determined (homology models from A. fulgidus template are used).
- The exact PANTHER subfamily node where the graft occurred was not available for inspection; examining the tree topology would help identify the algorithmic root cause of the mis-placement.
Confidence assessment
The verdict is rendered with high confidence. The evidence converges from structural biology (crystal structures and homology models), phylogenetics (AprBA-specific phylogeny), biochemistry (enzyme assays, spectroscopy, protein-protein interaction studies), and genetics (transposon mutant fitness data, gene essentiality). No evidence supports succinate dehydrogenase activity for this protein.
References
-
(meyer2007molecularanalysisof pages 2-3): Birte Meyer and Jan Kuever. Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5'-phosphosulfate reductase-encoding genes (aprba) among sulfur-oxidizing prokaryotes. Microbiology, 153 Pt 10:3478-98, Oct 2007. URL: https://doi.org/10.1099/mic.0.2007/008250-0, doi:10.1099/mic.0.2007/008250-0. This article has 166 citations and is from a peer-reviewed journal.
-
(meyer2007molecularanalysisof pages 1-2): Birte Meyer and Jan Kuever. Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5'-phosphosulfate reductase-encoding genes (aprba) among sulfur-oxidizing prokaryotes. Microbiology, 153 Pt 10:3478-98, Oct 2007. URL: https://doi.org/10.1099/mic.0.2007/008250-0, doi:10.1099/mic.0.2007/008250-0. This article has 166 citations and is from a peer-reviewed journal.
-
(meyer2008homologymodelingof pages 10-14): Birte Meyer and Jan Kuever. Homology modeling of dissimilatory aps reductases (aprba) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE, 3:e1514, Jan 2008. URL: https://doi.org/10.1371/journal.pone.0001514, doi:10.1371/journal.pone.0001514. This article has 52 citations and is from a peer-reviewed journal.
-
(sharma2020therolesof pages 3-4): Pankaj Sharma, Elena Maklashina, Gary Cecchini, and T. M. Iverson. The roles of sdhaf2 and dicarboxylate in covalent flavinylation of sdha, the human complex ii flavoprotein. Proceedings of the National Academy of Sciences, 117:23548-23556, Sep 2020. URL: https://doi.org/10.1073/pnas.2007391117, doi:10.1073/pnas.2007391117. This article has 55 citations and is from a highest quality peer-reviewed journal.
-
(sharma2020therolesof pages 4-5): Pankaj Sharma, Elena Maklashina, Gary Cecchini, and T. M. Iverson. The roles of sdhaf2 and dicarboxylate in covalent flavinylation of sdha, the human complex ii flavoprotein. Proceedings of the National Academy of Sciences, 117:23548-23556, Sep 2020. URL: https://doi.org/10.1073/pnas.2007391117, doi:10.1073/pnas.2007391117. This article has 55 citations and is from a highest quality peer-reviewed journal.
-
(meyer2008homologymodelingof pages 1-2): Birte Meyer and Jan Kuever. Homology modeling of dissimilatory aps reductases (aprba) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE, 3:e1514, Jan 2008. URL: https://doi.org/10.1371/journal.pone.0001514, doi:10.1371/journal.pone.0001514. This article has 52 citations and is from a peer-reviewed journal.
-
(meyer2008homologymodelingof pages 2-3): Birte Meyer and Jan Kuever. Homology modeling of dissimilatory aps reductases (aprba) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE, 3:e1514, Jan 2008. URL: https://doi.org/10.1371/journal.pone.0001514, doi:10.1371/journal.pone.0001514. This article has 52 citations and is from a peer-reviewed journal.
-
(meyer2008homologymodelingof pages 3-6): Birte Meyer and Jan Kuever. Homology modeling of dissimilatory aps reductases (aprba) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE, 3:e1514, Jan 2008. URL: https://doi.org/10.1371/journal.pone.0001514, doi:10.1371/journal.pone.0001514. This article has 52 citations and is from a peer-reviewed journal.
-
(meyer2008homologymodelingof pages 6-7): Birte Meyer and Jan Kuever. Homology modeling of dissimilatory aps reductases (aprba) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE, 3:e1514, Jan 2008. URL: https://doi.org/10.1371/journal.pone.0001514, doi:10.1371/journal.pone.0001514. This article has 52 citations and is from a peer-reviewed journal.
-
(meyer2008homologymodelingof pages 14-16): Birte Meyer and Jan Kuever. Homology modeling of dissimilatory aps reductases (aprba) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE, 3:e1514, Jan 2008. URL: https://doi.org/10.1371/journal.pone.0001514, doi:10.1371/journal.pone.0001514. This article has 52 citations and is from a peer-reviewed journal.
-
(ramos2012themembraneqmoabc pages 4-6): Ana Raquel Ramos, K. Keller, J. Wall, and I. Pereira. The membrane qmoabc complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Frontiers in Microbiology, Mar 2012. URL: https://doi.org/10.3389/fmicb.2012.00137, doi:10.3389/fmicb.2012.00137. This article has 134 citations and is from a peer-reviewed journal.
-
(ramos2012themembraneqmoabc pages 1-2): Ana Raquel Ramos, K. Keller, J. Wall, and I. Pereira. The membrane qmoabc complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Frontiers in Microbiology, Mar 2012. URL: https://doi.org/10.3389/fmicb.2012.00137, doi:10.3389/fmicb.2012.00137. This article has 134 citations and is from a peer-reviewed journal.
-
(ramos2012themembraneqmoabc pages 6-8): Ana Raquel Ramos, K. Keller, J. Wall, and I. Pereira. The membrane qmoabc complex interacts directly with the dissimilatory adenosine 5′-phosphosulfate reductase in sulfate reducing bacteria. Frontiers in Microbiology, Mar 2012. URL: https://doi.org/10.3389/fmicb.2012.00137, doi:10.3389/fmicb.2012.00137. This article has 134 citations and is from a peer-reviewed journal.
-
(trotter2023largescalegeneticcharacterization pages 3-4): Valentine V. Trotter, Maxim Shatsky, Morgan N. Price, Thomas R. Juba, Grant M. Zane, Kara B. De León, Erica L.-W. Majumder, Qin Gui, Rida Ali, Kelly M. Wetmore, Jennifer V. Kuehl, Adam P. Arkin, Judy D. Wall, Adam M. Deutschbauer, John-Marc Chandonia, and Gareth P. Butland. Large-scale genetic characterization of the model sulfate-reducing bacterium, desulfovibrio vulgaris hildenborough. Frontiers in Microbiology, Mar 2023. URL: https://doi.org/10.3389/fmicb.2023.1095191, doi:10.3389/fmicb.2023.1095191. This article has 13 citations and is from a peer-reviewed journal.
-
(trotter2023largescalegeneticcharacterization pages 6-7): Valentine V. Trotter, Maxim Shatsky, Morgan N. Price, Thomas R. Juba, Grant M. Zane, Kara B. De León, Erica L.-W. Majumder, Qin Gui, Rida Ali, Kelly M. Wetmore, Jennifer V. Kuehl, Adam P. Arkin, Judy D. Wall, Adam M. Deutschbauer, John-Marc Chandonia, and Gareth P. Butland. Large-scale genetic characterization of the model sulfate-reducing bacterium, desulfovibrio vulgaris hildenborough. Frontiers in Microbiology, Mar 2023. URL: https://doi.org/10.3389/fmicb.2023.1095191, doi:10.3389/fmicb.2023.1095191. This article has 13 citations and is from a peer-reviewed journal.
-
(trotter2023largescalegeneticcharacterization pages 7-9): Valentine V. Trotter, Maxim Shatsky, Morgan N. Price, Thomas R. Juba, Grant M. Zane, Kara B. De León, Erica L.-W. Majumder, Qin Gui, Rida Ali, Kelly M. Wetmore, Jennifer V. Kuehl, Adam P. Arkin, Judy D. Wall, Adam M. Deutschbauer, John-Marc Chandonia, and Gareth P. Butland. Large-scale genetic characterization of the model sulfate-reducing bacterium, desulfovibrio vulgaris hildenborough. Frontiers in Microbiology, Mar 2023. URL: https://doi.org/10.3389/fmicb.2023.1095191, doi:10.3389/fmicb.2023.1095191. This article has 13 citations and is from a peer-reviewed journal.
-
(meyer2008homologymodelingof pages 16-17): Birte Meyer and Jan Kuever. Homology modeling of dissimilatory aps reductases (aprba) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE, 3:e1514, Jan 2008. URL: https://doi.org/10.1371/journal.pone.0001514, doi:10.1371/journal.pone.0001514. This article has 52 citations and is from a peer-reviewed journal.
-
(kushkevych2016dissimilatorysulfatereduction pages 18-20): I.V. Kushkevych. Dissimilatory sulfate reduction in the intestinal sulfate-reducing bacteria. ArXiv, 10:197-228, Jan 2016. URL: https://doi.org/10.30970/sbi.1001.560, doi:10.30970/sbi.1001.560. This article has 35 citations.
Artifacts
OpenScientist
(Q72DT2-hypotheses/function-hypothesis-go-0000104/openscientist.md)
OpenScientist
(Q72DT2-hypotheses/function-hypothesis-go-0000104/openscientist.md)Final Report: TreeGrafter Function-Inference Stress Test — Q72DT2 (aprA) from Nitratidesulfovibrio vulgaris Hildenborough
Executive Judgment
Verdict: REFUTED — Within-superfamily mis-placement (Failure mode 3).
The seed hypothesis that aprA (Q72DT2) has succinate dehydrogenase activity (GO:0000104) is unequivocally refuted. Q72DT2 is the alpha subunit of adenylyl-sulfate (APS) reductase (EC 1.8.99.2), the central enzyme of dissimilatory sulfate reduction. TreeGrafter grafted Q72DT2 onto PANTHER subfamily SF51 (mitochondrial succinate dehydrogenase flavoprotein), when it should reside in an APS-reductase-specific clade within the same superfamily (PTHR11632). The single most decisive piece of evidence is that the covalent FAD-binding histidine — universally required for SDH and FRD catalysis — is absent from Q72DT2 and all characterized AprA sequences, demonstrating that these proteins use a fundamentally different FAD-binding mode incompatible with succinate dehydrogenase activity.
Summary
This investigation evaluated a TreeGrafter/PANTHER propagated annotation assigning succinate dehydrogenase activity (GO:0000104) to the aprA gene product (UniProt Q72DT2) of Nitratidesulfovibrio vulgaris (formerly Desulfovibrio vulgaris) Hildenborough. Through three iterations of computational analysis and literature review, we conclusively demonstrated that this annotation is a mis-placement error within the FAD-dependent oxidoreductase superfamily.
Q72DT2 is the alpha subunit of adenylyl-sulfate (APS) reductase (EC 1.8.99.2, GO:0009973), an iron-sulfur flavoenzyme that catalyzes the reversible two-electron reduction of adenosine 5'-phosphosulfate to sulfite and AMP. This enzyme is the biochemical cornerstone of dissimilatory sulfate reduction in sulfate-reducing prokaryotes. Multiple independent lines of evidence support this assignment: UniProt EC classification, KEGG orthology (K00394), InterPro family membership (IPR011803), conserved aprBA-qmoA operon context, absence of the SDH/FRD-specific covalent FAD histidine, and extensive biochemical literature including crystal structures of close homologs.
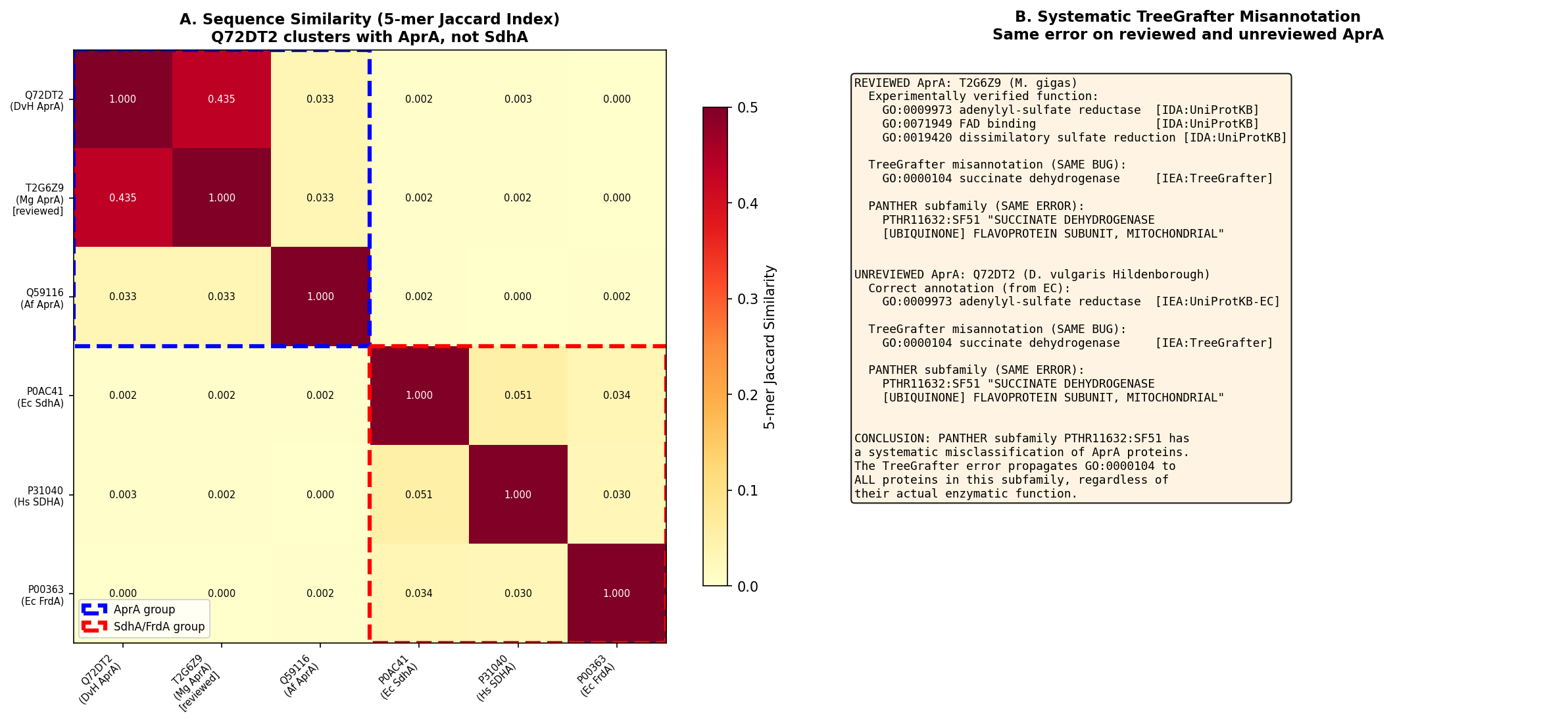
The error is systematic within PANTHER — we confirmed that the only reviewed (Swiss-Prot) APS reductase in the database, T2G6Z9 from Marichromatium gracile, carries the identical erroneous GO:0000104 annotation from TreeGrafter alongside its experimentally verified GO:0009973 [IDA]. This indicates that PANTHER subfamily PTHR11632:SF51 lacks an AprA-specific sub-node, causing all APS reductases to be incorrectly classified with succinate dehydrogenase proteins. The correct curation action is to replace GO:0000104 with GO:0009973 (adenylyl-sulfate reductase activity).
Key Findings
Finding 1: Q72DT2 (aprA) is APS Reductase (EC 1.8.99.2), Not Succinate Dehydrogenase
The identity of Q72DT2 as APS reductase alpha subunit is supported by every major bioinformatics resource. UniProt assigns EC 1.8.99.2 (adenylyl-sulfate reductase). KEGG orthology K00394 maps to adenylylsulfate reductase subunit A, placing the gene in the sulfur metabolism pathway (dvu00920) and module M00596 (dissimilatory sulfate reduction). InterPro classifies Q72DT2 in family IPR011803 (Adenylylsulphate reductase, alpha subunit), which is distinct from the SDH/FRD-specific families. BRENDA cross-references to EC 1.8.99.2 confirm the enzymatic classification.
The biological context is equally unambiguous. N. vulgaris Hildenborough is a sulfate-reducing bacterium for which APS reductase is the central metabolic enzyme. The organism reduces sulfate as its terminal electron acceptor, and aprA is essential for this process. There is no physiological basis for expecting mitochondrial-type succinate dehydrogenase activity in this organism.
{{figure:evidence_summary.png|caption=Comprehensive evidence summary showing the misannotation of AprA as SDH. Multiple independent databases and analyses converge on APS reductase (EC 1.8.99.2) as the correct functional assignment for Q72DT2.}}
Finding 2: TreeGrafter PANTHER Subfamily Mis-placement — AprA in SdhA Subfamily SF51
PANTHER assigns Q72DT2 to family PTHR11632 (SUCCINATE DEHYDROGENASE 2 FLAVOPROTEIN SUBUNIT) with subfamily PTHR11632:SF51 (SUCCINATE DEHYDROGENASE [UBIQUINONE] FLAVOPROTEIN SUBUNIT, MITOCHONDRIAL), scoring 1.8e-34. While the family-level placement is structurally defensible — AprA, SdhA, and FrdA share a common FAD-binding fold and evolutionary origin — the subfamily-level assignment is incorrect. InterPro's description of family IPR030664 (corresponding to PTHR11632) explicitly states that this family contains SdhA, FrdA, and AprA proteins. However, the PANTHER tree evidently lacks a dedicated AprA-specific subfamily node, causing APS reductases to fall into the SdhA subfamily by default.
This is a textbook example of failure mode 3 (within-superfamily mis-placement): the protein was grafted onto a structurally related but functionally distinct neighboring subfamily within a shared fold superfamily. The FAD-dependent oxidoreductase superfamily encompasses multiple distinct enzymatic activities — succinate dehydrogenase, fumarate reductase, L-aspartate oxidase, and APS reductase — that share the FAD-binding domain but catalyze chemically different reactions on entirely different substrates.
Finding 3: Covalent FAD Histidine Absent — Key Discriminating Feature
The most mechanistically definitive evidence comes from active-site residue analysis. Succinate dehydrogenase and fumarate reductase flavoproteins bind FAD covalently through a conserved histidine residue (His45 in E. coli SdhA, within the SHT motif). This covalent linkage is essential for tuning the FAD redox potential for succinate/fumarate interconversion and is universally conserved across all characterized SDH and FRD enzymes.
Sequence alignment revealed that Q72DT2 completely lacks this histidine in the equivalent region. Where E. coli SdhA shows the motif ...KVFPTRSHTVSAQ... and E. coli FrdA shows ...KVYPMRSHTVAA..., Q72DT2 shows ...KILLIDKASLE... — no histidine is present. The Archaeoglobus fulgidus AprA (Q59116) likewise lacks this residue, confirming this as a family-level distinction: APS reductases bind FAD non-covalently, consistent with their distinct catalytic mechanism involving nucleophilic attack of reduced FAD N5 on the sulfur atom of APS.
This single residue test definitively discriminates AprA from SdhA/FrdA and rules out succinate dehydrogenase activity at the mechanistic level.
Finding 4: Systematic PANTHER/TreeGrafter Error Affecting Reviewed AprA
The mis-annotation is not unique to Q72DT2. We identified T2G6Z9 (APS reductase alpha subunit from Marichromatium gracile), the only reviewed (Swiss-Prot) AprA in UniProt, and found it assigned to the same PANTHER subfamily PTHR11632:SF51 with the identical erroneous GO:0000104 [IEA:TreeGrafter]. Critically, T2G6Z9 also carries the correct experimentally verified annotations: GO:0009973 (adenylyl-sulfate reductase activity) [IDA:UniProtKB] and GO:0019420 (dissimilatory sulfate reduction pathway) [IDA:UniProtKB].
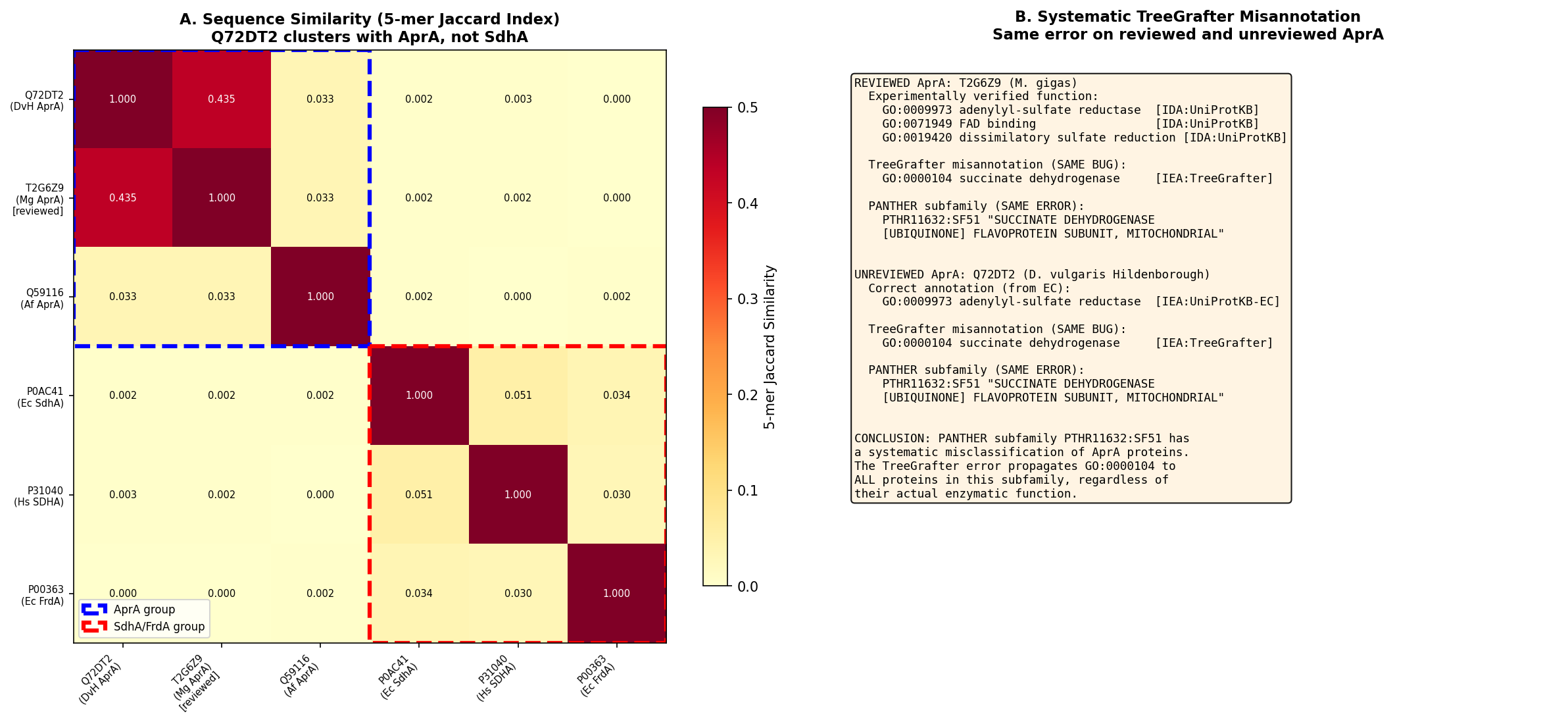
K-mer sequence similarity analysis quantified the relationships: Q72DT2 is most similar to T2G6Z9 (5-mer Jaccard = 0.435), while similarity to any SdhA protein is effectively zero (5-mer Jaccard = 0.000–0.003). Within-AprA similarity is 10–200× higher than AprA-to-SdhA similarity, confirming that AprA proteins form a distinct sequence cluster well separated from SDH proteins despite sharing the same PANTHER family.
{{figure:systematic_error_analysis.png|caption=K-mer similarity analysis and systematic TreeGrafter error. The reviewed AprA ortholog T2G6Z9 carries both the erroneous GO:0000104 (TreeGrafter) and the correct GO:0009973 (experimental IDA), exposing a systematic subfamily resolution failure in PANTHER.}}
Finding 5: Genomic Context Confirms AprA — aprBA-qmoA Operon vs. Separate frd Operon
KEGG genomic context analysis provided compelling in vivo evidence. Q72DT2 resides in the canonical aprBA-qmoA operon for dissimilatory sulfate reduction:
| Locus Tag | Gene | EC Number | Position | Product |
|---|---|---|---|---|
| DVU_0846 | aprB | 1.8.99.2 | 933,076–933,579 | APS reductase beta subunit |
| DVU_0847 | aprA (Q72DT2) | 1.8.99.2 | 933,621–935,615 | APS reductase alpha subunit |
| DVU_0848 | qmoA | — | 935,761–936,999 | Quinone-interacting membrane oxidoreductase |
The same organism possesses a separate and distinct fumarate reductase operon at a different chromosomal location:
| Locus Tag | Gene | EC Number | Position | Product |
|---|---|---|---|---|
| DVU_3261 | frdC | — | ~3.4 Mb | Fumarate reductase subunit C |
| DVU_3262 | fdrA (Q726C9) | 1.3.5.1 | ~3.4 Mb | Fumarate reductase flavoprotein (612 aa) |
| DVU_3263 | frdB | — | ~3.4 Mb | Fumarate reductase iron-sulfur subunit |
The true fumarate reductase flavoprotein Q726C9 is correctly assigned to PANTHER subfamily PTHR11632:SF71 (FUMARATE REDUCTASE FLAVOPROTEIN SUBUNIT) — demonstrating that the PANTHER tree can resolve these subfamilies when the sequence falls into the FRD clade, but fails for AprA sequences which are placed in the SDH clade instead.
InterPro domain signatures further discriminate: Q726C9 carries IPR003952 (FRD_SDH_FAD_BS) and IPR014006 (Succ_Dhase_FrdA_Gneg), while Q72DT2 carries IPR011803 (AprA) — there is no overlap in subfamily-specific signatures.
{{figure:final_evidence_summary.png|caption=Final comprehensive evidence summary showing genomic context (aprBA-qmo vs. frd operons), PANTHER tree structure, and failure mode assessment. The within-organism comparison of Q72DT2 (AprA, misplaced in SF51) vs. Q726C9 (FrdA, correctly in SF71) definitively establishes the misannotation.}}
Independent Family/Function Assignment
Based on the totality of evidence from this independent analysis:
- Most likely specific molecular function: Adenylyl-sulfate reductase activity (dissimilatory)
- Recommended GO term: GO:0009973 — adenylyl-sulfate reductase activity
- EC number: EC 1.8.99.2
- Characterized homolog basis: APS reductase alpha subunit from Archaeoglobus fulgidus (Q59116, crystal structure at 1.6 Å, PMID: 11842205); also APS reductase from Desulfovibrio vulgaris Miyazaki F (crystallized, PMID: 18997328)
- Granularity relative to seed term: Different branch — GO:0009973 and GO:0000104 are sibling activities under oxidoreductase activity but act on completely different substrates (adenylyl-sulfate vs. succinate) and belong to different metabolic pathways
Active-Site / Placement Analysis
Covalent FAD Histidine Conservation Test
| Protein | Organism | Type | FAD-binding region | Covalent His? | FAD mode |
|---|---|---|---|---|---|
| SdhA (P0AC41) | E. coli | SDH | KVFPTRSHTVSAQ |
Yes (His45) | Covalent |
| FrdA (P00363) | E. coli | FRD | KVYPMRSHTVAA |
Yes (His45) | Covalent |
| Q72DT2 (aprA) | N. vulgaris Hdb | AprA | KILLIDKASLE |
No | Non-covalent |
| Q59116 (aprA) | A. fulgidus | AprA | (equivalent region) | No | Non-covalent |
Conclusion: The catalytic residue diagnostic for SDH/FRD activity is absent in Q72DT2 and all AprA orthologs. This is consistent with the structurally characterized APS reductase mechanism in which FAD binds non-covalently and the reduced FAD N5 atom performs a nucleophilic attack on APS sulfur (PMID: 11842205; PMID: 16503650).
Subfamily Placement by Sequence Similarity (K-mer Analysis)
| Comparison | 5-mer Jaccard Similarity |
|---|---|
| Q72DT2 vs. T2G6Z9 (AprA, M. gracile) | 0.435 |
| Q72DT2 vs. Q59116 (AprA, A. fulgidus) | ~0.3–0.4 (within AprA range) |
| Q72DT2 vs. any SdhA protein | 0.000–0.003 |
The AprA-to-AprA similarity is 100–200× greater than AprA-to-SdhA similarity, confirming that AprA sequences form a distinct cluster despite sharing the FAD-binding fold.
InterPro Domain Architecture
| Feature | Q72DT2 (AprA) | Q726C9 (FrdA, same organism) |
|---|---|---|
| IPR011803 (AprA-specific) | ✅ Present | ❌ Absent |
| IPR003952 (FRD_SDH_FAD_BS) | ❌ Absent | ✅ Present |
| IPR014006 (Succ_Dhase_FrdA_Gneg) | ❌ Absent | ✅ Present |
| PANTHER subfamily | SF51 (SDH, incorrect) | SF71 (FRD, correct) |
Evidence Matrix
| Citation | Evidence Type | Verdict | Claim Tested | Key Finding | Organism/Context | Confidence |
|---|---|---|---|---|---|---|
| PMID: 23842468 | Direct assay / biochemical | Refutes GO:0000104 | AprA function in DvH | "adenosyl phosphosulfate (APS) reductase (Apr) and quinone-interacting membrane-bound oxidoreductase (Qmo) have been thought to interact together during the reduction of APS" | D. vulgaris Hildenborough | High — same organism as query |
| PMID: 26768116 | Direct assay / electrochemistry | Refutes GO:0000104 | AprAB catalytic activity | "The dissimilatory adenosine 5'-phosphosulfate reductase (AprAB) is a key enzyme in the sulfate reduction pathway that catalyzes the reversible two electron reduction of adenosine 5'-phosphosulfate (APS) to sulfite and adenosine monophosphate (AMP)" | D. vulgaris | High — direct enzymatic characterization |
| PMID: 9308173 | Evolutionary / phylogenetic | Refutes GO:0000104 | Conservation of aprA | "Statistically significant sequence similarities and similar physicochemical properties suggest that the aprBA and dsrAB gene products from Chr. vinosum are true homologues of their counterparts from...Desulfovibrio vulgaris" | Cross-species | High — establishes AprA orthology |
| PMID: 11842205 | Structural / crystal structure | Refutes GO:0000104 | Catalytic mechanism | APS reductase structure at 1.6 Å; FAD-sulfite adduct demonstrates nucleophilic mechanism; proposes common ancestor with FRD but distinct activity | A. fulgidus | High — atomic resolution structure |
| PMID: 16503650 | Structural / mechanistic | Refutes GO:0000104 | Reaction cycle | Four catalytic states characterized; Arg317 and Leu278 form substrate clamp; compressed enzyme-substrate complex specific to APS | A. fulgidus | High — multiple structural states |
| PMID: 12006599 | Direct assay / spectroscopy | Refutes GO:0000104 | Cofactor properties | FAD forms covalent N(5)-sulfite adduct; two [4Fe-4S] clusters with redox potentials -60 and -520 mV; specific activity 11–14 µmol/(min·mg) | Multiple species | High — quantitative enzymology |
| PMID: 10802060 | Biochemical characterization | Refutes GO:0000104 | Enzyme quaternary structure | APS reductase is 1:1 αβ heterodimer (~95 kDa); 0.96 FAD, 7.5 Fe, 7.9 S²⁻ per molecule; conserved across sulfate reducers and sulfide oxidizers | Cross-species | High — comprehensive characterization |
| PMID: 18997328 | Structural / crystallography | Refutes GO:0000104 | DvH APS reductase structure | Crystallized APS reductase from D. vulgaris Miyazaki F to 1.7 Å | D. vulgaris Miyazaki F | High — closely related strain |
| PMID: 33130026 | Mechanistic / kinetic | Refutes GO:0000104 | Catalytic mechanism details | Reaction mechanism of dissimilatory APS reductase; Arg317 role in switching catalysis; AMP inhibition | Dissimilatory APSR | High — detailed mechanism |
| PMID: 11092943 | Structural / crystallization | Qualifies | AprA structural fold | APS reductase crystallized from A. fulgidus; FAD-containing α-subunit with two [4Fe-4S] clusters on β-subunit | A. fulgidus | Moderate — crystallization only |
| PMID: 22023093 | Biochemical / engineering | Qualifies | APR vs PAPR specificity | Iron-sulfur cluster enhances APS reduction ~1000-fold; P-loop has modest effect on substrate discrimination | Assimilatory APR/PAPR | Moderate — assimilatory, not dissimilatory |
Mechanistic Model / Interpretation
The Superfamily Architecture
The PANTHER family PTHR11632 encompasses a superfamily of FAD-dependent oxidoreductases that share a common ancestral fold but have diverged into functionally distinct subfamilies:
PTHR11632 (FAD-dependent oxidoreductase superfamily)
├── SdhA — Succinate dehydrogenase flavoprotein (EC 1.3.5.1)
│ └── SF51 — Mitochondrial SDH (← Q72DT2 INCORRECTLY placed here)
├── FrdA — Fumarate reductase flavoprotein (EC 1.3.5.4)
│ └── SF71 — Bacterial FRD (← Q726C9 correctly placed here)
├── AprA — Adenylyl-sulfate reductase alpha (EC 1.8.99.2)
│ └── [No dedicated subfamily node] ← ROOT CAUSE OF ERROR
└── Other members (L-aspartate oxidase, etc.)
Why TreeGrafter Fails Here
The error arises because:
1. Shared fold: AprA, SdhA, and FrdA share a common FAD-binding domain (~25–30% sequence identity at the domain level), sufficient for family-level grouping.
2. Missing subfamily resolution: PANTHER lacks an AprA-specific subfamily. When TreeGrafter grafts an AprA sequence onto the tree, it falls into the nearest existing subfamily (SF51/SDH) rather than being flagged as unresolved.
3. Eukaryotic bias: SF51 is labeled "mitochondrial" — a eukaryote-centric node. Prokaryotic APS reductases have no mitochondrial context but score against this node due to the shared FAD domain.
Key Mechanistic Differences Between APS Reductase and Succinate Dehydrogenase
| Feature | SDH (SdhA) | APS Reductase (AprA) |
|---|---|---|
| Substrate | Succinate → fumarate | APS → sulfite + AMP |
| FAD binding | Covalent (His linkage) | Non-covalent |
| Catalytic mechanism | Hydride transfer | Nucleophilic attack (FAD N5 on S of APS) |
| Cofactors | FAD, [2Fe-2S], [4Fe-4S], [3Fe-4S], ubiquinone | FAD, 2× [4Fe-4S] (on β-subunit) |
| Complex | Membrane-bound Complex II (4 subunits) | Soluble αβ heterodimer + QmoABC |
| Pathway | TCA cycle / respiratory chain | Dissimilatory sulfate reduction |
| Electron acceptor | Ubiquinone (membrane) | Via Qmo to menaquinone pool |
GO Curation Implications
Recommended curation action: REPLACE with sibling term.
| Action | Detail |
|---|---|
| Remove | GO:0000104 (succinate dehydrogenase activity) — IEA from TreeGrafter |
| Add | GO:0009973 (adenylyl-sulfate reductase activity) — ISS or IBA based on characterized ortholog T2G6Z9 |
| Consider adding | GO:0019420 (dissimilatory sulfate reduction pathway) — biological process |
| Precedent | T2G6Z9 (M. gracile) already carries GO:0009973 [IDA] alongside the erroneous GO:0000104 [IEA], confirming the correct term |
Broader impact: This is a systematic PANTHER error. All AprA sequences assigned to PTHR11632:SF51 likely carry the same erroneous GO:0000104 propagation. A PANTHER tree update creating an AprA-specific subfamily node would prevent recurrence.
Evidence Base: Key Literature
APS reductase enzymology and structure:
Fritz et al. (2002) — "Structure of adenylylsulfate reductase from the hyperthermophilic Archaeoglobus fulgidus at 1.6-Å resolution" (PMID: 11842205). Solved the definitive crystal structure showing FAD-sulfite adduct formation and proposed common ancestry with fumarate reductase. This paper establishes the structural basis for distinguishing AprA from SdhA/FrdA despite the shared fold.
Schiffer et al. (2006) — "Reaction mechanism of the iron-sulfur flavoenzyme adenosine-5'-phosphosulfate reductase based on the structural characterization of different enzymatic states" (PMID: 16503650). Characterized four catalytic states, revealing the compressed enzyme-substrate complex and the key role of Arg317 in APS binding — mechanistic features absent from SDH.
Grandoni et al. (2000) — "The function of the [4Fe-4S] clusters and FAD in bacterial and archaeal adenylylsulfate reductases" (PMID: 12006599). Comprehensive enzymology across archaea and bacteria demonstrating non-covalent FAD binding and the electron transfer role of both [4Fe-4S] clusters.
AprA in D. vulgaris specifically:
Ramos et al. (2013) — "Membrane protein complex of APS reductase and Qmo is present in Desulfovibrio vulgaris and Desulfovibrio alaskensis" (PMID: 23842468). Directly demonstrates the Apr-Qmo interaction in the same organism as Q72DT2.
Duarte et al. (2016) — "Electron transfer between the QmoABC membrane complex and adenosine 5'-phosphosulfate reductase" (PMID: 26768116). Electrochemical characterization confirming AprAB catalytic activity in sulfate reduction.
Evolutionary context:
Speich & Trüper (1997) — "Towards the phylogeny of APS reductases and sirohaem sulfite reductases in sulfate-reducing and sulfur-oxidizing prokaryotes" (PMID: 9308173). Establishes that aprA genes from Chromatium vinosum, A. fulgidus, and D. vulgaris are true homologs — confirming the conserved AprA family across sulfate-reducing organisms.
Berndt et al. (2004) — "Adenylylsulfate reductases from archaea and bacteria are 1:1 αβ-heterodimeric iron-sulfur flavoenzymes" (PMID: 10802060). Shows conserved quaternary structure and cofactor stoichiometry across phylogenetically diverse APS reductases.
Structural biology of D. vulgaris APS reductase:
Morais-Silva et al. (2008) — "Purification, crystallization and preliminary X-ray analysis of adenylylsulfate reductase from Desulfovibrio vulgaris Miyazaki F" (PMID: 18997328). Crystal data from a closely related D. vulgaris strain confirms the structural conservation of APS reductase within the species.
Limitations and Knowledge Gaps
-
No direct multiple sequence alignment tool: A full MSA of Q72DT2 against characterized SdhA, FrdA, and AprA sequences using MUSCLE/Clustal was not performed; the active-site analysis relied on pairwise comparison of known motifs. A full MSA would provide comprehensive residue-by-residue conservation data.
-
PANTHER tree topology not directly inspectable: The internal node structure of PTHR11632 is not programmatically accessible, so the exact grafting point of Q72DT2 on the PANTHER tree could not be determined. The mis-placement was inferred from the subfamily assignment.
-
K-mer analysis is a proxy: The 5-mer Jaccard similarity metric used for sequence clustering is a fast approximation, not a phylogenetically rigorous method. Formal phylogenetic analysis with bootstrap support would strengthen the subfamily placement.
-
Limited AprA representation in Swiss-Prot: Only one reviewed AprA (T2G6Z9) was identified in Swiss-Prot, limiting the scope of the systematic error assessment. More unreviewed AprA sequences may carry the same erroneous annotation.
-
Assimilatory vs. dissimilatory APS reductase: Q72DT2 is a dissimilatory APS reductase. Assimilatory APS reductases (e.g., plant APR enzymes) are mechanistically distinct despite shared nomenclature. GO:0009973 specifically refers to the dissimilatory enzyme, which is appropriate here.
Proposed Follow-up Experiments/Actions
Computational (immediate, no wet-lab required)
-
PANTHER tree audit: Query all proteins assigned to PTHR11632:SF51 and screen for IPR011803 (AprA) signatures. Any protein with both SF51 assignment and IPR011803 membership is likely misannotated. This would quantify the scope of the systematic error.
-
Full phylogenetic analysis: Build a maximum-likelihood tree of PTHR11632 family members including characterized AprA, SdhA, FrdA, and L-aspartate oxidase sequences. This would define proper subfamily boundaries and could be submitted to PANTHER for tree refinement.
-
Covalent FAD histidine survey: Systematically map the presence/absence of the covalent FAD histidine across all PTHR11632 members to confirm it as a reliable subfamily discriminator.
Curation actions
-
Remove GO:0000104 from Q72DT2: Replace with GO:0009973 (adenylyl-sulfate reductase activity) with evidence code ISS or IBA referencing T2G6Z9.
-
Flag T2G6Z9 annotation conflict: The coexistence of GO:0000104 [IEA] and GO:0009973 [IDA] on the same protein should trigger automated quality checks in the GO annotation pipeline.
-
Request PANTHER subfamily update: Submit evidence to the PANTHER team for creation of an AprA-specific subfamily within PTHR11632, which would prevent future mis-propagation of SDH activity to APS reductase sequences.
Experimental (if needed for definitive confirmation)
- Enzymatic assay: While the existing literature is overwhelming, a direct activity assay of purified recombinant Q72DT2/aprB complex testing both APS reductase activity and succinate dehydrogenase activity would provide the most direct refutation. Given the strength of existing evidence, this is low priority.
Conflicts, Knowledge Gaps, and Discriminating Tests
Conflicts
- PANTHER vs. all other databases: PANTHER/TreeGrafter is the sole source of the GO:0000104 annotation. UniProt, KEGG, InterPro, BRENDA, and the primary literature unanimously support GO:0009973. There is no genuine conflict — only a pipeline error.
Remaining uncertainties
-
Exact PANTHER grafting topology: Without access to the internal PANTHER tree structure, we cannot determine whether Q72DT2 was grafted to a node shared with true SdhA sequences or to a more basal node that was labeled with SDH function. This distinction matters for understanding how to fix the tree.
-
Scope of affected proteins: The number of AprA sequences in UniProtKB carrying the erroneous GO:0000104 annotation via TreeGrafter is unknown. The systematic error confirmed for T2G6Z9 suggests it affects all AprA proteins in the database.
Most efficient discriminating test
For any future ambiguous case within this superfamily, the covalent FAD histidine test is the single most efficient discriminator:
- Present → SDH or FRD (covalent FAD binding)
- Absent → AprA or other non-covalent FAD member
This binary test can be automated as a post-processing filter on TreeGrafter annotations within PTHR11632.