Deep Research
Falcon
(atg16-deep-research-falcon.md)
Falcon
(atg16-deep-research-falcon.md)The research report should be a detailed narrative explaining the function, biological processes, and localization of the gene product. Citations should be given for all claims.
You should prioritize authoritative reviews and primary scientific literature when conducting research. You can supplement
this with annotations you find in gene/protein databases, but these can be outdated or inaccurate.
We are specifically interested in the primary function of the gene - for enzymes, what reaction is catalyzed, and what is the substrate specificity? For transporters, what is the substrate? For structural proteins or adapters, what is the broader structural role? For signaling molecules, what is the role in the pathway.
We are interested in where in or outside the cell the gene product carries out its function.
We are also interested in the signaling or biochemical pathways in which the gene functions. We are less interested in broad pleiotropic effects, except where these elucidate the precise role.
Include evidence where possible. We are interested in both experimental evidence as well as inference from structure, evolution, or bioinformatic analysis. Precise studies should be prioritized over high-throughput, where available.
Research Report: Functional Annotation of atg16 (UniProt O94656; ORF SPBC405.05) in Schizosaccharomyces pombe (strain 972)
0) Executive summary (identity verified)
The target protein is Atg16 from fission yeast (Schizosaccharomyces pombe strain 972), encoded by ORF SPBC405.05 and matching UniProt accession O94656. Primary S. pombe evidence explicitly assigns SPBC405.05 to atg16 and shows it encodes an Atg16-family component of the autophagy conjugation machinery with a conserved Atg5-binding N-terminus and C-terminal coiled-coil typical of Atg16 proteins (https://doi.org/10.1371/journal.pgen.1003715; published 2013-08) (sun2013globalanalysisof pages 3-5, sun2013globalanalysisof pages 14-15).
1) Key concepts and definitions (current understanding)
1.1 Macroautophagy and Atg8/LC3 lipidation
Macroautophagy (“autophagy”) is a conserved pathway in which cytoplasmic material is sequestered into a double-membrane autophagosome and delivered to the vacuole/lysosome for degradation. A central molecular hallmark is lipidation of Atg8 (LC3/GABARAP in mammals), i.e., covalent conjugation of Atg8 to the membrane lipid phosphatidylethanolamine (PE), generating Atg8–PE on forming autophagic membranes (xu2022fissionyeastautophagy pages 2-4, noda2023structuralbiologyof pages 1-4).
1.2 The ATG12–ATG5–ATG16 “E3-like” complex
Atg8 lipidation proceeds via a ubiquitin-like cascade: Atg7 (E1-like) activates Atg8, transfers it to Atg3 (E2-like), and an E3-like complex—the Atg12–Atg5–Atg16 complex—stimulates transfer of Atg8 onto PE and helps determine where lipidation occurs (xu2022fissionyeastautophagy pages 2-4, noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 1-4). In this system, Atg16 is not the catalytic enzyme, but a scaffolding/targeting subunit critical for organizing the E3-like machinery on the correct membrane surface (noda2023structuralbiologyof pages 8-12, nishimura2020emergingrolesof pages 8-10).
2) Molecular function of S. pombe Atg16 (what it does)
2.1 Primary functional role: enabling Atg8–PE formation at autophagic membranes
In S. pombe, Atg16 functions as a core component of the Atg12–Atg5–Atg16 module required for productive autophagy, as demonstrated by the CFP-Atg8 processing assay: atg16Δ cells are defective in CFP-Atg8 processing under nitrogen starvation (https://doi.org/10.1371/journal.pgen.1003715; 2013-08) (sun2013globalanalysisof pages 3-5, sun2013globalanalysisof media 4581e895). Because CFP/GFP fusions are relatively protease-resistant and release free fluorophore upon vacuolar delivery, this processing assay is widely used as a bulk-autophagy flux readout in fission yeast (xu2022fissionyeastautophagy pages 2-4).
2.2 Protein–protein interactions: Atg16 binds Atg5 in vivo
A key biochemical feature of Atg16 is its association with Atg5. In S. pombe, Atg16-YFP co-immunoprecipitates with Atg5-Myc and this interaction is observed both in wild-type and in atg12Δ cells, supporting that Atg16 can bind Atg5 independently of Atg12 conjugation (https://doi.org/10.1371/journal.pgen.1003715; 2013-08) (sun2013globalanalysisof pages 3-5, sun2013globalanalysisof media 4581e895). This aligns with the conserved Atg16 architecture: an N-terminal Atg5-binding region and a coiled-coil region that supports higher-order assembly (noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 1-4).
3) Subcellular localization and where Atg16 acts
3.1 Localization to the PAS/pre-autophagosomal structure during starvation
Microscopy in S. pombe shows Atg16-YFP colocalizing with CFP-Atg8 at punctate structures corresponding to the PAS/pre-autophagosomal structure during starvation (sun2013globalanalysisof media 4581e895). This supports that Atg16 acts at early autophagosome biogenesis sites, consistent with its role in organizing Atg8 lipidation machinery on the forming phagophore (xu2022fissionyeastautophagy pages 5-7, noda2023structuralbiologyof pages 1-4).
3.2 Quantitative dynamics: PAS puncta lifetimes
Time-lapse analysis of Atg8 puncta in wild-type fission yeast indicates PAS structures are dynamic on the order of minutes, with puncta lifetimes reported mostly ~100–200 seconds in the cited study (sun2013globalanalysisof pages 3-5). This provides a useful temporal scale for Atg16-dependent recruitment and conjugation events.
4) Pathway placement and S. pombe-specific mechanistic features
4.1 Atg18a-dependent recruitment of the Atg12–Atg5–Atg16 complex (distinctive feature)
A prominent S. pombe mechanistic detail is the non-redundant role of Atg18 paralogs in organizing the conjugation machinery: Atg18a is uniquely required to target the Atg12–Atg5·Atg16 complex to the PAS, supporting Atg8 conjugation to PE (sun2013globalanalysisof pages 10-11, xu2022fissionyeastautophagy pages 5-7). Consistent with this, microscopy shows Atg5/Atg16 puncta are lost in atg18aΔ cells, and Atg8 puncta formation is also impaired when either atg16 or atg18a is deleted (sun2013globalanalysisof media 4581e895).
This Atg18a-dependent targeting was a central conclusion of the original S. pombe study that identified Atg16 as a core Atg factor (https://doi.org/10.1371/journal.pgen.1003715; 2013-08) (sun2013globalanalysisof pages 10-11, sun2013globalanalysisof media 4581e895).
4.2 Conserved vs. divergent recruitment logic across eukaryotes
Recent mechanistic synthesis emphasizes that across eukaryotes, Atg16/ATG16L1 recruitment to PI3P-positive autophagic membranes often involves PI3P-binding PROPPIN/WIPI proteins. In budding yeast, Atg21 binds PI3P and engages the Atg16 coiled-coil; in mammals, WIPI2 similarly recruits ATG16L1 (noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 15-17). A 2023 primary study further shows that coiled-coil-mediated dimerization of Atg16 is required for binding Atg21, highlighting the importance of the Atg16 coiled-coil oligomeric state for PROPPIN-mediated recruitment (https://doi.org/10.1098/rsob.230192; published 2023-11) (buenoarribas2023coiledcoilmediateddimerizationof pages 1-2).
While these PROPPIN/WIPI recruitment mechanisms were studied outside S. pombe, they provide a strong comparative framework: S. pombe uses an Atg18-family protein (Atg18a) as a critical determinant for recruiting/positioning the Atg12–Atg5–Atg16 complex at the PAS (sun2013globalanalysisof pages 10-11, xu2022fissionyeastautophagy pages 5-7).
5) Mutant phenotypes and experimental evidence (S. pombe)
5.1 Autophagy flux defect (CFP-Atg8 processing)
Deletion of atg16 produces a loss-of-function autophagy phenotype as assessed by CFP-Atg8 processing after nitrogen starvation (samples taken before and 8 h after shift to nitrogen-free medium in the cited work) (sun2013globalanalysisof pages 3-5). Figure panels retrieved from the primary paper visually document the processing defect and related localization phenotypes (sun2013globalanalysisof media 4581e895).
5.2 PAS assembly phenotype (Atg8 puncta)
Microscopy evidence shows atg16Δ cells fail to generate normal Atg8 puncta at the PAS during starvation; similar absence of puncta is seen in atg18aΔ, consistent with Atg18a’s role in recruiting the Atg12–Atg5–Atg16 complex (sun2013globalanalysisof media 4581e895).
5.3 Genetic/physiological context
Fission yeast core autophagy genes are important for survival during nitrogen starvation and for developmental programs (mating/sporulation). The fission-yeast autophagy machinery review summarizes that core atg deletions disrupt viability under starvation and cause mating/sporulation defects, consistent with atg16 being in the core set (xu2022fissionyeastautophagy pages 2-4).
6) Recent developments (prioritizing 2023–2024)
6.1 2023 structural biology synthesis: architecture and recruitment interfaces
A 2023 review focused on structural biology of the Atg8/Atg12 conjugation systems highlights:
- Atg16 contains an Atg5-binding N-terminus and a parallel coiled-coil that mediates homodimerization; a structural length scale of the coiled-coil dimer is reported (~13 nm) (https://doi.org/10.1080/27694127.2023.2277582; published 2023-11) (noda2023structuralbiologyof pages 8-12).
- The Atg12–Atg5–Atg16 complex functions as an E3-like scaffold that can stimulate Atg8/LC3 lipidation, in part by engaging Atg3 and promoting lipidation at the correct membrane site (noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 15-17).
- Recruitment to PI3P-positive membranes can be mediated by PROPPIN/WIPI family adaptors (noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 15-17).
These developments provide an updated, expert-level mechanistic context for interpreting Atg16 function in yeast, including S. pombe, even when S. pombe-specific high-resolution structural data are limited (noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 15-17).
6.2 2024 literature in the retrieved set
Within the retrieved corpus, 2024 content was primarily broader autophagy reviews (e.g., microautophagy) rather than Atg16-specific S. pombe mechanistic work; thus, for Atg16 itself, the most directly relevant recent advances captured here are in 2023 structural/recruitment understanding (noda2023structuralbiologyof pages 8-12, buenoarribas2023coiledcoilmediateddimerizationof pages 1-2).
7) Current applications and real-world implementations
7.1 Functional genomics discovery pipeline in S. pombe
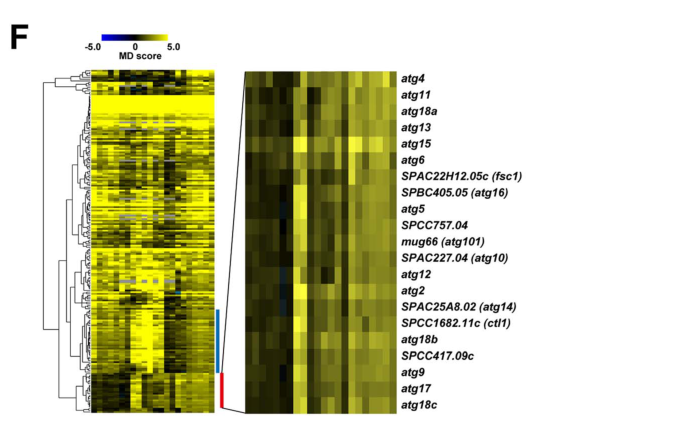
Atg16 was discovered/validated in a large-scale, real-world functional-genomics workflow: a genome-wide barcode sequencing mating-defect screen in S. pombe that screened 2,915 deletion mutants and generated 63,146 MD-score measurements across 22 screens, calling 206 hits at FDR < 0.1 in all three standard-condition screens (https://doi.org/10.1371/journal.pgen.1003715; 2013-08) (sun2013globalanalysisof pages 2-3, sun2013globalanalysisof pages 1-2). This study expanded experimentally defined S. pombe autophagy factors from 14 to 23 (sun2013globalanalysisof pages 3-5).
These statistics are useful for interpreting confidence in atg16’s assignment as a core autophagy gene and for benchmarking future screens (sun2013globalanalysisof pages 2-3).
7.2 Standard assays that operationalize Atg16 function
In fission yeast, Atg16 function is routinely operationalized via:
- CFP/GFP/YFP-Atg8 processing assays (bulk autophagy flux) (xu2022fissionyeastautophagy pages 2-4).
- PAS localization microscopy with fluorescently tagged Atg factors (Atg16-YFP with Atg8 puncta) (sun2013globalanalysisof media 4581e895).
These are “real-world” implementations in cell biology labs because Atg16 is a key node whose deletion produces clear readouts (flux and PAS-assembly defects) (sun2013globalanalysisof pages 3-5, sun2013globalanalysisof media 4581e895).
8) Expert opinions and authoritative analysis (from reviews)
The fission yeast autophagy machinery review (Cells, 2022-03; https://doi.org/10.3390/cells11071086) presents Atg16 as part of the core conjugation system and emphasizes that S. pombe has both conserved machinery and meaningful differences from S. cerevisiae, including differential roles among Atg18 paralogs and PAS recruitment logic for the Atg12–Atg5·Atg16 complex (xu2022fissionyeastautophagy pages 2-4, xu2022fissionyeastautophagy pages 5-7). The 2023 structural biology review frames Atg16 as a central E3-platform organizer and clarifies modern consensus on how recruitment interfaces (coiled-coil, PROPPIN/WIPI interactions) tune where Atg8 lipidation occurs (noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 15-17).
9) Evidence-focused summary table
The following table consolidates the strongest annotation statements, evidence types, and quantitative/statistical data captured here.
| Category | Key findings (concise) | Evidence (study + year) | URL/DOI | Notes/quantitative data |
|---|---|---|---|---|
| Identity/domains | UniProt target O94656 matches S. pombe ORF SPBC405.05, named atg16; the protein is homologous to budding-yeast Atg16 and contains an N-terminal Atg5-binding region plus a C-terminal coiled-coil domain. (sun2013globalanalysisof pages 3-5, sun2013globalanalysisof pages 14-15) | Sun et al., 2013; Xu & Du, 2022 | https://doi.org/10.1371/journal.pgen.1003715; https://doi.org/10.3390/cells11071086 | Sequence/domain assignment is from the S. pombe primary paper; aligns with UniProt family call. |
| Molecular function | Atg16 is the scaffold subunit of the Atg12–Atg5–Atg16 complex, which functions as the E3-like factor for Atg8 conjugation to phosphatidylethanolamine (PE) and helps determine where Atg8 lipidation occurs on autophagic membranes. (xu2022fissionyeastautophagy pages 2-4, xu2022fissionyeastautophagy pages 5-7, noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 1-4) | Xu & Du, 2022; Noda, 2023 | https://doi.org/10.3390/cells11071086; https://doi.org/10.1080/27694127.2023.2277582 | Not an enzyme with its own catalytic active site; functions as an adaptor/scaffold within the E3-like complex. |
| Complex/partners | In S. pombe, Atg16 physically associates with Atg5; co-immunoprecipitation detected the Atg5–Atg16 interaction both in wild type and atg12Δ cells, indicating Atg5 binding does not strictly require Atg12. Atg18a also functionally/physically links to the complex for PAS targeting. (sun2013globalanalysisof pages 3-5, sun2013globalanalysisof pages 10-11, sun2013globalanalysisof pages 11-13) | Sun et al., 2013 | https://doi.org/10.1371/journal.pgen.1003715 | Co-IP inputs/IPs reported in the paper include 1% input / 20% IP for one assay setup (sun2013globalanalysisof pages 3-5). |
| Localization | Atg16 localizes to the PAS (phagophore assembly site / pre-autophagosomal structure) during starvation and colocalizes with CFP-Atg8; proper PAS localization of the Atg12–Atg5–Atg16 complex requires Atg18a. (sun2013globalanalysisof pages 10-11, xu2022fissionyeastautophagy pages 5-7, sun2013globalanalysisof media 4581e895) | Sun et al., 2013; Xu & Du, 2022 | https://doi.org/10.1371/journal.pgen.1003715; https://doi.org/10.3390/cells11071086 | Microscopy figures show Atg16-YFP, Atg5-YFP, and Atg18a-YFP colocalizing with CFP-Atg8; in atg18aΔ, Atg5/Atg16 puncta are lost (sun2013globalanalysisof media 4581e895). |
| Phenotypes/assays | atg16Δ is defective in CFP-Atg8 processing, supporting a requirement for bulk autophagy; atg16Δ cells also fail to form normal Atg8 puncta at the PAS in the cited imaging analysis. (sun2013globalanalysisof pages 3-5, sun2013globalanalysisof media 4581e895) | Sun et al., 2013 | https://doi.org/10.1371/journal.pgen.1003715 | Time course for the CFP-Atg8 assay included samples before and 8 h after nitrogen starvation; the snippet is qualitative and does not report a numeric processing fraction for atg16Δ (sun2013globalanalysisof pages 3-5). |
| S. pombe-specific notes | A notable fission-yeast-specific feature is that Atg18a is uniquely required to recruit the Atg12–Atg5–Atg16 complex to the PAS; the three Atg18/WIPI paralogs are not redundant, and Atg18a differs functionally from Atg18b/c. (sun2013globalanalysisof pages 10-11, xu2022fissionyeastautophagy pages 5-7, sun2013globalanalysisof pages 11-13) | Sun et al., 2013; Xu & Du, 2022 | https://doi.org/10.1371/journal.pgen.1003715; https://doi.org/10.3390/cells11071086 | In the cited phenotypes, atg18aΔ abolishes Atg8 puncta, whereas atg18bΔ/atg18cΔ increase Atg8 puncta, consistent with different pathway roles (sun2013globalanalysisof pages 10-11). |
| Recent (2023-2024) mechanistic context | Recent reviews/primary studies reinforce that Atg16/ATG16L1 has an Atg5-binding N-terminus and a dimeric coiled-coil that recruits the E3-like complex through PROPPIN/WIPI proteins; Atg16 dimerization is important for PROPPIN binding, and the coiled-coil has been described as about 13 nm long in structural context. (noda2023structuralbiologyof pages 8-12, noda2023structuralbiologyof pages 15-17, buenoarribas2023coiledcoilmediateddimerizationof pages 1-2, nishimura2020emergingrolesof pages 8-10) | Noda, 2023; Bueno-Arribas et al., 2023 | https://doi.org/10.1080/27694127.2023.2277582; https://doi.org/10.1098/rsob.230192 | These recent papers are mostly broader mechanistic/structural context, not S. pombe-specific biochemistry for O94656 alone. |
| Applications/statistics | S. pombe Atg16 was discovered/validated through a genome-wide mating-phenotype barcode screen and is now part of the tractable fission-yeast autophagy toolkit using CFP/GFP-Atg8 processing assays. The screen analyzed 2,915 deletion mutants, generated 63,146 MD-score measurements across 22 screens, and called 206 hits at FDR < 0.1 in all three standard screens; the study expanded the experimentally defined S. pombe autophagy factor set from 14 to 23 genes. (sun2013globalanalysisof pages 13-14, sun2013globalanalysisof pages 3-5, sun2013globalanalysisof pages 2-3, sun2013globalanalysisof pages 1-2, sun2013globalanalysisof pages 14-15) | Sun et al., 2013; Xu & Du, 2022 | https://doi.org/10.1371/journal.pgen.1003715; https://doi.org/10.3390/cells11071086 | Practical implementation details include >99% spore purity, barcode filters requiring ≥12 reads and ≥1/40 upper-quartile counts, and use of CFP/GFP-Atg8 processing as routine autophagy-flux assays (sun2013globalanalysisof pages 13-14, xu2022fissionyeastautophagy pages 2-4). |
Table: This table summarizes the core functional annotation of Schizosaccharomyces pombe Atg16 (UniProt O94656 / SPBC405.05), including identity, molecular role, interaction partners, localization, phenotypes, and recent mechanistic context. It is useful as a compact evidence map tying S. pombe-specific findings to broader 2023–2024 autophagy research.
10) Key figures (visual evidence)
The primary S. pombe study contains figure panels showing: defective CFP-Atg8 processing in atg16Δ, PAS colocalization of Atg16-YFP with CFP-Atg8, and Atg18a-dependent PAS targeting/co-IP evidence for recruitment logic (sun2013globalanalysisof media 4581e895).
11) Limitations and evidence gaps (for completeness)
- In the retrieved evidence, S. pombe Atg16 phenotypes are well supported qualitatively (flux defect, PAS localization) but explicit numeric flux values (e.g., densitometry of free CFP/GFP) and autophagosome size measurements for atg16Δ were not present in the provided text snippets; addressing these would require deeper extraction from full paper sections or additional S. pombe primary literature focused on ultrastructure/quantification.
- Most 2023–2024 advances in the retrieved set concern general/conserved conjugation-system mechanism and recruitment (structural biology and PROPPIN/WIPI interactions), rather than S. pombe–exclusive biochemical innovations.
References
-
(sun2013globalanalysisof pages 3-5): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
-
(sun2013globalanalysisof pages 14-15): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
-
(xu2022fissionyeastautophagy pages 2-4): Dan-Dan Xu and Li-Lin Du. Fission yeast autophagy machinery. Cells, 11:1086, Mar 2022. URL: https://doi.org/10.3390/cells11071086, doi:10.3390/cells11071086. This article has 29 citations.
-
(noda2023structuralbiologyof pages 1-4): Nobuo N. Noda. Structural biology of the atg8 and atg12 conjugation systems. Autophagy Reports, Nov 2023. URL: https://doi.org/10.1080/27694127.2023.2277582, doi:10.1080/27694127.2023.2277582. This article has 12 citations.
-
(noda2023structuralbiologyof pages 8-12): Nobuo N. Noda. Structural biology of the atg8 and atg12 conjugation systems. Autophagy Reports, Nov 2023. URL: https://doi.org/10.1080/27694127.2023.2277582, doi:10.1080/27694127.2023.2277582. This article has 12 citations.
-
(nishimura2020emergingrolesof pages 8-10): Taki Nishimura and Sharon A. Tooze. Emerging roles of atg proteins and membrane lipids in autophagosome formation. Cell Discovery, May 2020. URL: https://doi.org/10.1038/s41421-020-0161-3, doi:10.1038/s41421-020-0161-3. This article has 346 citations and is from a peer-reviewed journal.
-
(sun2013globalanalysisof media 4581e895): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
-
(xu2022fissionyeastautophagy pages 5-7): Dan-Dan Xu and Li-Lin Du. Fission yeast autophagy machinery. Cells, 11:1086, Mar 2022. URL: https://doi.org/10.3390/cells11071086, doi:10.3390/cells11071086. This article has 29 citations.
-
(sun2013globalanalysisof pages 10-11): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
-
(noda2023structuralbiologyof pages 15-17): Nobuo N. Noda. Structural biology of the atg8 and atg12 conjugation systems. Autophagy Reports, Nov 2023. URL: https://doi.org/10.1080/27694127.2023.2277582, doi:10.1080/27694127.2023.2277582. This article has 12 citations.
-
(buenoarribas2023coiledcoilmediateddimerizationof pages 1-2): Miranda Bueno-Arribas, Celia Cruz-Cuevas, María-Angeles Navas, Ricardo Escalante, and Olivier Vincent. Coiled-coil-mediated dimerization of atg16 is required for binding to the proppin atg21. Open Biology, Nov 2023. URL: https://doi.org/10.1098/rsob.230192, doi:10.1098/rsob.230192. This article has 5 citations and is from a peer-reviewed journal.
-
(sun2013globalanalysisof pages 2-3): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
-
(sun2013globalanalysisof pages 1-2): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
-
(sun2013globalanalysisof pages 11-13): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
-
(sun2013globalanalysisof pages 13-14): Ling-Ling Sun, Ming Li, Fang Suo, Xiao-Man Liu, En-Zhi Shen, Bing Yang, Meng-Qiu Dong, Wan-Zhong He, and Li-Lin Du. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genetics, 9:e1003715, Aug 2013. URL: https://doi.org/10.1371/journal.pgen.1003715, doi:10.1371/journal.pgen.1003715. This article has 123 citations and is from a domain leading peer-reviewed journal.
Artifacts
Citations
- xu2022fissionyeastautophagy pages 2-4
- sun2013globalanalysisof pages 3-5
- buenoarribas2023coiledcoilmediateddimerizationof pages 1-2
- noda2023structuralbiologyof pages 8-12
- sun2013globalanalysisof pages 2-3
- sun2013globalanalysisof pages 10-11
- sun2013globalanalysisof pages 14-15
- noda2023structuralbiologyof pages 1-4
- nishimura2020emergingrolesof pages 8-10
- xu2022fissionyeastautophagy pages 5-7
- noda2023structuralbiologyof pages 15-17
- sun2013globalanalysisof pages 1-2
- sun2013globalanalysisof pages 11-13
- sun2013globalanalysisof pages 13-14
- https://doi.org/10.1371/journal.pgen.1003715;
- https://doi.org/10.1098/rsob.230192;
- https://doi.org/10.1080/27694127.2023.2277582;
- https://doi.org/10.3390/cells11071086
- https://doi.org/10.3390/cells11071086;
- https://doi.org/10.1080/27694127.2023.2277582
- https://doi.org/10.1371/journal.pgen.1003715
- https://doi.org/10.1098/rsob.230192
- https://doi.org/10.1371/journal.pgen.1003715,
- https://doi.org/10.3390/cells11071086,
- https://doi.org/10.1080/27694127.2023.2277582,
- https://doi.org/10.1038/s41421-020-0161-3,
- https://doi.org/10.1098/rsob.230192,
Perplexity
(atg16-deep-research-perplexity.md)
Perplexity
(atg16-deep-research-perplexity.md)Comprehensive Functional Analysis of the Autophagy-Related Protein 16 (ATG16) from Schizosaccharomyces pombe
The autophagy-related protein 16 (ATG16), encoded by the atg16 gene in Schizosaccharomyces pombe (fission yeast, UniProt accession O94656), serves as a critical structural and catalytic scaffold protein in the autophagy pathway. This protein forms an essential complex with ATG12 and ATG5 that functions as an E3-like ubiquitin ligase to facilitate the conjugation of the ubiquitin-like protein ATG8 to phosphatidylethanolamine during autophagosome biogenesis. Beyond its canonical role in autophagy, ATG16 participates in membrane tethering, proteasome-autophagy crosstalk, and non-canonical autophagy pathways. The protein's localization to the pre-autophagosomal structure (PAS) and its dynamic interactions with various regulatory proteins enable precise spatiotemporal control of autophagosome formation. This report synthesizes current knowledge of ATG16 function, structure, and mechanisms of action, with emphasis on experimental evidence and evolutionary conservation.
Protein Identity and Gene Characterization
The atg16 gene in Schizosaccharomyces pombe encodes autophagy protein 16, a conserved component of the autophagy machinery[1][4]. The gene is identified with the systematic designation SPBC405.05 and produces a protein with UniProt accession O94656[1][4]. The fission yeast ATG16 represents an evolutionarily distant homolog of well-characterized mammalian ATG16L1 and budding yeast (Saccharomyces cerevisiae) Atg16, with the yeast proteins sharing fundamental structural organization and functional properties that have been conserved across eukaryotes[19][38]. Notably, S. pombe is estimated to have diverged from S. cerevisiae more than 500 million years ago, yet autophagy factors have remained substantially conserved, underscoring the ancient and essential nature of this cellular pathway[19].
The atg16 gene was originally identified through genome-wide screens designed to uncover autophagy-related genes in fission yeast[19]. Prior to systematic characterization, three core autophagy components—Atg10, Atg14, and Atg16—had not been identified in S. pombe, despite being well-characterized in budding yeast[19]. The discovery of these genes completed the roster of expected core autophagy machinery components in fission yeast and permitted comprehensive characterization of the autophagy system across a diverse yeast species[19]. The identification of Atg16 in fission yeast was particularly significant because it demonstrated that this protein family represents a genuinely conserved component of autophagosome biogenesis machinery across evolutionarily distant organisms.
Structural Organization and Domain Architecture
The ATG16 protein exhibits a modular domain architecture that has been preserved throughout eukaryotic evolution[10][21][35]. The fission yeast Atg16 protein, like its mammalian counterpart ATG16L1, contains an N-terminal domain responsible for binding to the ATG12–ATG5 conjugate, followed by a coiled-coil domain essential for protein oligomerization[35]. In mammals, ATG16L1 additionally contains a C-terminal WD40 domain composed of seven repeats of the WD40 motif, though this domain is absent in yeast Atg16 proteins[24][35][43]. The core Atg5-binding motif, termed the AFIM (Atg5-interacting motif), adopts an alpha-helical conformation that mediates specific recognition by the ATG5 protein[24]. Crystal structural analysis of yeast Atg5 complexed with the N-terminal region of Atg16 revealed that the N-terminal region of Atg16 exhibits a helical structure and binds within a groove formed by the ubiquitin-like domains and helix-rich regions of Atg5[10][24].
The coiled-coil domain of Atg16 functions in mediating protein-protein interactions essential for complex assembly. This domain exhibits coiled-coil propensity and contains conserved leucine residues that mediate homodimerization through a leucine zipper-like mechanism[16][32]. The coiled-coil architecture is crucial because ATG16 must form higher-order oligomeric complexes with the ATG12–ATG5 conjugate to execute its full biological function[21]. In yeast, the Atg16 protein forms a homodimer that further associates with a single Atg12–Atg5 conjugate to generate an asymmetric heteromeric complex[10]. The functional significance of Atg16 homodimerization has been demonstrated through mutagenic disruption of the leucine zipper, which prevents dimer formation and causes complete loss of autophagy function despite normal expression and localization of the protein[16][32].
The membrane-binding capability of Atg16 derives from an amphipathic alpha helix located in the central region of the protein (approximately amino acid residues 113–131 in yeast, based on structural analysis of related proteins)[16][32]. This amphipathic helix exhibits a strong hydrophobic face that inserts into lipid bilayers, thereby anchoring the protein to membrane structures[16][32]. The distinct structural regions of Atg16—the Atg5-binding domain at the N-terminus, the leucine zipper dimerization domain, and the amphipathic membrane-binding helix—represent functionally independent modules that can be separated experimentally to define specific roles[16][32]. In fission yeast, these structural features are preserved, suggesting that the S. pombe Atg16 protein maintains the essential functional architecture required for autophagosome biogenesis across eukaryotic cells.
The ATG12–ATG5–ATG16 Complex: Assembly and Stoichiometry
The ATG12–ATG5–ATG16 complex represents one of the most critical assemblies in the autophagy pathway, functioning as a specialized E3-like ubiquitin ligase[2][8][9][27]. This complex is assembled through sequential conjugation reactions beginning with the covalent attachment of ATG12 to ATG5, catalyzed by the E1-like enzyme ATG7 and the E2-like enzyme ATG10[9][27][39]. The resulting ATG12–ATG5 conjugate then associates non-covalently with ATG16, which is recruited to this complex through its N-terminal AFIM domain[4][10][25]. The assembly process has been characterized in detail through biochemical reconstitution studies and structural analysis, revealing that all three components are required to generate a functional E3 ligase capable of promoting ATG8 lipidation[8][9].
The stoichiometry of the ATG12–ATG5–ATG16 complex has been determined through multiple experimental approaches including gel filtration chromatography, cross-linking mass spectrometry, and crystallographic analysis[10][24][25]. In mammalian cells, the complex forms a large oligomeric assembly with an apparent molecular mass of approximately 800 kilodaltons[24][43][55]. This size is consistent with a complex composed of multiple copies of the ATG12–ATG5 conjugate associated with a homodimeric or higher oligomeric form of ATG16L1[24][43][55]. In yeast, Atg16 forms a homodimer that associates with the Atg12–Atg5 conjugate to generate approximately a 350 kilodalton complex[4][21]. The difference in stoichiometry between mammalian and yeast systems reflects variations in ATG16 oligomerization capacity, as mammalian ATG16L1 can form higher-order multimers more readily than yeast Atg16[24].
The assembly of this complex is essential for autophagy function, as demonstrated by the observation that mutations disrupting any interface within the complex—whether between ATG12 and ATG5, between ATG5 and ATG16, or within the Atg16 homodimer—completely abolish autophagy[16][32][34][43]. Furthermore, the spatial organization of the complex is important; the three-dimensional arrangement of the ATG12–ATG5–ATG16 assembly determines its ability to position substrates correctly for the lipidation reaction[25][29]. The coiled-coil domain and homodimeric organization of ATG16 appear to provide multiple membrane-binding surfaces, potentially allowing one ATG16 dimer to engage multiple membrane locations and thereby facilitate membrane tethering and vesicle clustering[31][32].
Recruitment to the Pre-Autophagosomal Structure: Dual Pathways
ATG16 must be recruited to the site of autophagosome biogenesis—the pre-autophagosomal structure (PAS)—to execute its catalytic and scaffolding functions. Recent research has revealed that this recruitment occurs through two distinct, partially redundant mechanisms, each contributing specific functions to autophagy initiation[17][28][34][51][54]. The first mechanism operates through phosphatidylinositol 3-phosphate (PI3P) binding and involves the WIPI proteins (WD repeat domain phosphoinositide-interacting proteins, also known as Atg18 in yeast)[7][37][38]. The second mechanism is less well-characterized but involves direct interaction between the ATG16 complex and the ATG1 kinase complex through the N-terminal region of ATG12[17][28][34][51][54].
The PI3P-dependent recruitment pathway is initiated when the class III phosphatidylinositol 3-kinase (PI3K) complex produces PI3P at nascent autophagosomal membranes[7][37][38]. In mammalian cells, the WIPI2 protein binds directly to the WIPI2-interacting region (W2IR) of ATG16L1, which spans approximately residues 207–230[7][13]. Crystal structural analysis of the WIPI2–ATG16L1 complex revealed that the W2IR adopts an alpha-helical conformation and binds within an electropositive and hydrophobic groove located between beta-propeller blades 2 and 3 of WIPI2[7][13]. This interaction is highly specific; mutations at the WIPI2 binding interface significantly reduce or block the recruitment of the ATG12–ATG5–ATG16L1 complex to synthetic membranes in vitro and impair LC3 lipidation[7][13]. In fission yeast, the multiple Atg18/WIPI proteins—Atg18a, Atg18b, and Atg18c—play distinct roles in autophagy, with Atg18a being uniquely required for the targeting of the Atg12–Atg5·Atg16 complex[38]. Atg18a appears to serve as a binding platform for the recruitment of the Atg12–Atg5·Atg16 complex to the PAS in S. pombe[38].
The second recruitment mechanism discovered through recent research demonstrates that the Atg16 complex interacts with the Atg1 kinase complex via the N-terminal region of Atg12[17][28][34][51][54]. This interaction was demonstrated through co-immunoprecipitation experiments showing that the Atg16 complex, which requires intact Atg5 and Atg12 to form, physically associates with components of the Atg1 complex including Atg17[17][28][34]. The N-terminal 56 amino acids of Atg12 are specifically required for this interaction; deletion of this region (Atg12ΔN56) completely abolishes binding to the Atg1 complex while leaving the conjugation of Atg12 to Atg5 and the interaction with Atg16 intact[17][28][34]. The Atg1 complex serves as a scaffold for PAS organization and appears to represent the earliest-acting complex in autophagosome biogenesis; therefore, this Atg12-dependent recruitment mechanism is proposed to represent a PI3P-independent, early targeting pathway that precedes the PI3P-dependent pathway mediated by Atg21/WIPI proteins[17][28][34][51][54].
Importantly, these two recruitment pathways exhibit functional redundancy such that disruption of either pathway alone results in only partial autophagy defects, but simultaneous disruption of both pathways completely abolishes PAS localization of the Atg16 complex and prevents autophagy[17][28][34][51][54]. When cells or yeast strains lack both Atg21 and the Atg12 N-terminal region, the Atg16 complex cannot localize to the PAS and autophagy is completely blocked[17][28][34]. This redundancy ensures robust autophagosome formation under diverse cellular conditions and provides flexibility in the timing of Atg16 recruitment relative to other autophagy machinery components[17][28][34][51][54].
Membrane Binding and Tethering: Molecular Mechanisms
ATG16 functions as a peripheral membrane-binding protein through its amphipathic alpha helix and contributes to membrane organization and vesicle clustering during autophagosome biogenesis. The membrane-binding characteristics of ATG16 have been dissected through subcellular fractionation experiments, in vitro liposome sedimentation assays, and reconstitution studies using giant unilamellar vesicles[8][11][16][31][32]. These studies reveal that ATG16, when in complex with ATG5 and ATG12, efficiently binds to lipid membranes, while the ATG12–ATG5 conjugate alone exhibits severely reduced membrane binding[8][11][31]. This observation indicates that ATG16 activation of membrane binding by ATG5 is a fundamental regulatory mechanism—ATG16 association with the ATG12–ATG5 conjugate unmasks a membrane-binding surface on ATG5 that is otherwise occluded by ATG12 conjugation[8][11][31].
The mechanism by which ATG16 activates membrane binding appears to involve both direct membrane interaction and relief of inhibition by ATG12[8][11][16][31][32]. When ATG16 associates with the ATG12–ATG5 conjugate through its N-terminal AFIM domain, a conformational change occurs that exposes hydrophobic residues on ATG5 required for lipid interaction[8][11][31][32]. ATG16 itself contributes to membrane binding through its amphipathic helix, and since ATG16 forms a homodimer, this dimerization may provide multiple membrane-binding surfaces or create a more extensive lipid-binding interface[16][31][32]. In vitro studies using giant unilamellar vesicles demonstrate that the ATG12–ATG5–ATG16 complex causes massive aggregation and clustering of vesicles in a manner that is entirely dependent on ATG16[8][11][31]. The clustering and tethering function of ATG16 appears to be mediated through the formation of bridges between membrane surfaces, as the homodimeric nature of ATG16 would allow a single dimer to contact multiple lipid bilayers simultaneously[8][31][32].
The functional significance of membrane binding by ATG16 is demonstrated by the observation that mutations preventing membrane association (such as the K160E/R171E double mutant of yeast Atg5, or disruption of the amphipathic helix in Atg16) result in severe autophagy defects despite normal PAS recruitment[8][11][31]. Cells expressing these membrane-binding-deficient mutants fail to efficiently promote ATG8 lipidation and show impaired formation of autophagosomes[8][11][31]. Furthermore, membrane binding is essential for the delivery of the ATG8-loaded E2 enzyme ATG3 to the lipid substrate phosphatidylethanolamine, suggesting that the ATG16 complex functions as a scaffold that brings together the catalytic machinery (Atg7, Atg3, and the lipid substrate) on the membrane surface[8][31][49]. This brings the substrate into proximity with the catalytic machinery, thereby greatly accelerating the lipidation reaction and specifying the location where ATG8 conjugation occurs[8][25][31][49].
Catalytic Function in the ATG8 Lipidation Cascade
The primary catalytic role of the ATG16 complex involves facilitating the conjugation of the ubiquitin-like protein ATG8 to phosphatidylethanolamine (PE), a critical lipidation reaction essential for autophagosome biogenesis[2][9][25][27][49]. This reaction proceeds through a series of sequential enzymatic steps analogous to ubiquitin conjugation, except that the ultimate target is a lipid rather than a protein lysine residue[9][27][49]. The pathway involves four key enzymatic activities: the E1-like enzyme ATG7, which activates ATG8; the E2-like enzyme ATG3, which receives ATG8 via a thioester bond; the E3-like enzyme complex ATG12–ATG5–ATG16, which catalyzes the final transfer; and specific lipases or deconjugases that reverse the reaction when needed[9][27][30][49].
The ATG12–ATG5–ATG16 complex functions as an E3-like enzyme analogous to ubiquitin ligases, but with a critical difference: rather than catalyzing peptide bond formation between substrate and target lysine as in ubiquitination, it catalyzes ester bond formation between the carboxyl terminus of ATG8 and the primary amino group of the PE headgroup[9][27][49][52]. Biochemical reconstitution of this reaction using purified components and synthetic liposomes demonstrates that the E3 complex is absolutely required for efficient ATG8 lipidation[8][9][11][25]. The E3 complex enhances the catalytic activity of the E2 enzyme ATG3 through allosteric mechanisms; in the absence of E3, ATG3 exhibits very low activity in transferring ATG8 to PE, but when the full E3 complex is present, the lipidation reaction is dramatically accelerated[8][11][25][49].
The molecular mechanism by which the E3 complex enhances E2 activity has been clarified through structural and biochemical studies. The ATG12 component of the E3 complex directly interacts with ATG3 and positions this E2 enzyme optimally relative to the PE substrate on the membrane[25][49]. The ATG5 component provides membrane-binding activity that brings the entire catalytic assembly to the membrane surface where the PE substrate resides[8][11][31]. The ATG16 component amplifies membrane binding through its homodimeric organization and amphipathic helix[16][31][32]. Furthermore, ATG16 may function in specifying the site of ATG8 lipidation by clustering membrane regions and ensuring that ATG8 conjugation occurs at autophagy-relevant membranes rather than throughout the cell[25][29].
Recent structural and biophysical studies have revealed that multiple members of the ATG8/LC3-GABARAP family are substrates for this lipidation pathway in mammalian cells[29][52]. The efficiency of lipidation varies among these different ATG8 homologs, with GABARAP subfamily proteins generally being conjugated more efficiently than LC3 subfamily proteins when studied in vitro[29]. The E3 complex increases and speeds up lipidation of all family members and also increases the capacity of lipidated ATG8 family proteins to induce vesicle tethering and fusion[29]. However, interestingly, the E3 complex appears to restrict the fusion-promoting activity of lipidated ATG8 proteins, suggesting that the E3 may facilitate controlled membrane expansion during the elongation phase of autophagosome biogenesis by promoting tethering while constraining premature fusion[29].
Autophagy-Independent Functions and Crosstalk Pathways
Although ATG16 is best characterized for its role in canonical autophagy, emerging evidence indicates that this protein participates in autophagy-independent cellular functions and mediates crosstalk between autophagy and other cellular degradation pathways[2][15][20]. The ubiquitin proteasome system (UPS) and autophagy represent the two major cellular degradation pathways; growing evidence suggests that ATG16 and other autophagy machinery proteins regulate the balance between these systems and even mediate their functional coordination[2][15][36].
One important autophagy-independent function of ATG16 involves LC3-associated phagocytosis (LAP), a non-canonical autophagy pathway that occurs downstream of phagocytosis and does not require core autophagy initiation factors[15][20]. LAP has been shown to require the WD40 domain of mammalian ATG16L1, but not factors required for canonical autophagy such as ULK1, AMBRA1, ATG14, or FIP200[15][20]. This compartmentalization of function demonstrates that the WD40 domain of ATG16L1 is a critical module for non-canonical autophagy functions distinct from canonical autophagosome biogenesis[20]. The WD40 domain serves to recruit ATG16L1 to single-membrane endolysosomal compartments rather than double-membrane autophagosomes, representing a novel membrane recruitment mechanism distinct from the WIPI2/PI3P-dependent recruitment to canonical autophagosomes[20].
ATG16 also participates in crosstalk with the ubiquitin proteasome system through its interaction with the selective autophagy adaptor p62/SQSTM1 and its role in proteaphagy—the autophagic degradation of proteasomes themselves[2][15][36]. In plants, proteaphagy has been demonstrated to involve ATG16 homologs and appears to be mediated by direct recognition of proteasomal subunits through interaction with autophagy machinery[2]. The selective autophagy adaptor p62/SQSTM1 acts as a bridge between the ubiquitinated cargo destined for proteasomal degradation and the autophagy machinery, and this process appears to require the ATG16-containing complex[2][36].
The relationship between autophagy and the proteasome system is bidirectional; inhibition of autophagy leads to accumulation of p62 and impaired clearance of ubiquitinated proteins through the proteasome, despite proteasomal catalytic activity remaining normal[2][36]. This suggests that autophagy, and specifically ATG16-dependent processes, facilitate delivery of substrates to the proteasome or prevent sequestration of these substrates in aggregates that are inaccessible to proteasomal degradation[2][36]. The p62-mediated targeting of ubiquitinated protein aggregates to autophagy depends on interaction with LC3/ATG8 through the LC3-interacting region (LIR) domain of p62, and the ability of p62 to interact with the ubiquitin proteasome complex suggests that ATG16-containing autophagosomal membranes may preferentially sequence certain UPS substrates[2][36].
Cellular Localization and Subcellular Dynamics
The subcellular localization of ATG16 undergoes dynamic changes during the autophagy cycle, reflecting its recruitment to and dissociation from sites of autophagosome biogenesis. In vegetative (non-starved) cells, ATG16 is predominantly cytosolic, with only a small fraction localized to the perivacuolar PAS[41]. Upon induction of autophagy through nutrient starvation, ATG16 rapidly forms punctate structures that colocalize with other Atg proteins, particularly ATG8 and ATG5[38][41]. These puncta represent the PAS, where multiple autophagy factors concentrate to drive autophagosome biogenesis[14][41][57]. Quantitative analysis of autophagy protein stoichiometry at the PAS in yeast reveals that ATG16 is present at relatively low abundance compared to other Atg proteins such as ATG8, suggesting that ATG16 may function at limiting concentrations[41].
The dynamic localization of ATG16 to punctate structures is absolutely dependent on the presence of other autophagy machinery components[17][28][34][38]. In S. pombe, deletion of atg18a (encoding the WIPI protein) blocks PAS recruitment of both Atg5 and Atg16, preventing the formation of autophagosomal structures[38]. Similarly, disruption of the Atg12-dependent recruitment pathway through deletion of the N-terminal region of Atg12 prevents PAS localization of Atg16[17][28][34]. This strict dependence on recruitment mechanisms ensures that ATG16 localizes only to sites where autophagosome formation is occurring and where the proper lipid and protein substrates are assembled[17][28][34][38].
Following the nucleation and initial expansion of the phagophore, ATG16 is thought to remain associated with the expanding membrane, but this has not been directly visualized in most systems. The timing of ATG16 association with autophagosomal membranes appears to be relatively transient; ATG16 is recruited early in phagophore biogenesis during the nucleation and expansion phases but may be released as the phagophore matures and closes to form a sealed autophagosome[25][41]. This transient association is consistent with the reversible nature of ATG8 lipidation, which is catalyzed by the ATG16-containing E3 complex but can be reversed by deconjugating enzymes[30]. The recycling of ATG16 and other autophagy machinery proteins from completed autophagosomes back to cytosolic pools or to the PAS for reuse represents an important aspect of autophagy flux[30][41].
The membrane dynamics and localization of ATG16 are regulated by post-translational modifications including phosphorylation[2][9][27]. In mammalian cells, the ATG12–ATG5–ATG16L1 complex can be phosphorylated at specific residues, and these modifications appear to regulate both its recruitment to autophagosomes and its catalytic activity[2]. In yeast, phosphorylation of Ser139 in ATG12 promotes complex formation, whereas methylation of Lys151 in ATG16 inhibits complex assembly in cardiomyocytes[2]. These regulatory modifications provide additional layers of control over ATG16 function, allowing cells to modulate autophagy flux in response to distinct environmental signals and metabolic states.
Structural Basis of ATG16 Function and Evolutionary Conservation
The structural features of ATG16 that mediate its biological functions have been conserved throughout eukaryotic evolution, from single-celled yeasts to multicellular organisms[10][19][24][35]. Comparative structural analysis of Atg16 proteins from S. cerevisiae, S. pombe, and mammals reveals conservation of the N-terminal Atg5-binding domain, the central coiled-coil dimerization domain, and in mammals, the C-terminal WD40 repeats[10][24][35][43]. The AFIM motif responsible for Atg5 binding is particularly well-conserved, with key residues such as Arg-35 and Phe-46 (in yeast Atg16) being absolutely critical for both Atg5 interaction and autophagy function[10].
The crystal structure of the yeast Atg5–Atg16(1-46) complex at 1.97 Angstrom resolution reveals the atomic basis of Atg5-Atg16 interaction[10]. The structure shows that Atg5 comprises two ubiquitin-like domains flanking a helix-rich domain; the N-terminal region of Atg16 binds within a groove formed by these three domains of Atg5[10]. In vitro analysis demonstrated that Arg-35 and Phe-46 of Atg16 are crucial for this interaction, and Atg16 mutants with changes at these residues fail to localize to the PAS and cannot restore autophagy in Atg16-deficient cells[10]. Furthermore, these critical Atg16 residues are positioned such that they form hydrophobic and electrostatic interactions with complementary surfaces on Atg5, explaining the high specificity of this interaction[10].
The coiled-coil domain of ATG16, which mediates homodimerization, has been subject to detailed structural and functional analysis[16][32][35]. Crystallographic studies of the isolated coiled-coil domain (amino acids 55–142 in yeast Atg16) demonstrate that this region forms a parallel coiled-coil dimer stabilized by hydrophobic interactions between leucine residues at the "a" and "d" positions of the heptad repeat pattern characteristic of coiled-coils[16][32]. Mutation of critical leucine residues (L71A, L75A, L85A, L99A) that mediate the coiled-coil interaction prevents dimer formation and completely abolishes autophagy function, demonstrating the biological necessity of Atg16 homodimerization[16][32].
The membrane-binding amphipathic helix of ATG16 (approximately residues 113–131 in yeast) contains a characteristic pattern of hydrophobic and polar residues consistent with insertion into lipid bilayers[16][32]. The hydrophobic face of this helix comprises hydrophobic amino acids (leucines and isoleucines) that favorably interact with the acyl chains of membrane lipids, while the polar face contains charged and polar residues that interact with the aqueous environment and lipid headgroups[16][32]. This amphipathic organization allows ATG16 to stably anchor to membranes without forming a transmembrane helix; instead, it functions as a peripheral membrane protein[16][32]. The critical importance of this domain is demonstrated by point mutations that disrupt membrane binding, which result in loss of autophagy function despite normal expression, localization to the PAS, and binding to Atg5[8][11][16][31][32].
Disease Associations and Functional Variants
The ATG16L1 gene has achieved clinical significance through its association with Crohn's disease, a chronic inflammatory bowel disease affecting millions of people worldwide[44][47]. Multiple genome-wide association studies have identified the Thr300Ala (T300A) polymorphism in ATG16L1 as one of the most frequently associated genetic variants with Crohn's disease susceptibility[44][47]. The T300A mutation results in enhanced degradation of ATG16L1 via caspase-3-mediated cleavage, leading to reduced steady-state levels of ATG16L1 protein and decreased autophagic flux[44][47]. This reduced autophagy impairs several antimicrobial and immune functions, including diminished clearance of intracellular bacteria and altered secretion of antimicrobial peptides by intestinal Paneth cells[44][47].
Studies in genetically modified mice have revealed that the T300A variant affects both gut microbial composition and immune cell populations in a manner consistent with impaired autophagy[44]. Mice expressing the T300A allele display alterations in bacterial abundance, with increases in specific bacterial species including Bacteroides ovatus[44]. Furthermore, these mice show elevated Th17 and Th1 T cell populations in the gut-associated lymphoid tissue, suggesting enhanced pro-inflammatory immune responses[44]. These changes occur before overt disease symptoms develop, indicating that the autophagy defect associated with T300A creates a predisposing condition for inflammatory disease[44]. The functional consequence of reduced ATG16L1 abundance is impaired bacterial sequestration through xenophagy and altered innate immune responses to microbial products[44].
The structural basis for the association of T300A with disease susceptibility has been clarified through biochemical studies demonstrating that this position is a caspase-3 cleavage site[44]. During cellular stress or certain inflammatory responses, activated caspase-3 cleaves ATG16L1 at the T300A position, releasing the C-terminal WD40 domain from the remainder of the protein[44]. The resulting protein fragments exhibit reduced ability to promote autophagy and inflammatory responses, creating a molecular basis for the observed disease phenotype[44]. The T300A mutation enhances the rate of caspase-3-mediated cleavage relative to the wild-type threonine, explaining why the polymorphism represents a loss-of-function variant[44].
Beyond the T300A variant, other disease associations have been identified for ATG16L2, a mammalian-specific isoform of ATG16L[2][43][55]. A study examining ATG16L2 expression in multiple sclerosis (MS) patients found that both mRNA and protein levels of ATG16L2 were decreased in MS patient samples compared to healthy controls[2]. This observation suggests that ATG16L2 may play an important role in autophagy of T cells and may serve as a potential biomarker for predicting relapse rates in MS patients, although further investigation is required[2]. The identification of disease associations with ATG16 variants and isoforms underscores the fundamental importance of this protein in immune homeostasis and inflammatory regulation.
Evolutionary Perspectives and Comparative Biology
The conservation of ATG16 across eukaryotic organisms from single-celled yeasts to mammals reflects the ancient origin and essential nature of autophagy in cellular homeostasis[19][38][39][46]. Systematic comparative analysis of autophagy factors across yeast species, including S. cerevisiae and S. pombe, reveals that core autophagy components including ATG16 have been maintained with high fidelity across evolutionary time despite 500 million years of divergence[19][38]. The evolutionary conservation extends to the organization of the ATG12–ATG5–ATG16 complex and the fundamental mechanisms of autophagosome biogenesis, suggesting that these processes represent evolutionarily optimal solutions to the cellular problem of protein degradation and recycling[19][38][39].
Comparative studies between yeast and mammalian autophagy have revealed both conserved and divergent aspects of ATG16 function[19][38]. While the basic structural features and biochemical activities of ATG16 are highly conserved, mammalian systems have evolved additional complexity through the presence of multiple isoforms (ATG16L1 and ATG16L2) and the WD40 domain present only in mammals[43][55]. The WD40 domain of ATG16L1 enables distinct functions including participation in non-canonical autophagy processes, suggesting that mammalian evolution has elaborated the basic yeast ATG16 scaffold to perform additional cellular tasks[20][43]. The identification of ATG16 and other autophagy factors in ancient lineages such as plants and protists indicates that autophagy predates the diversification of eukaryotes and likely arose as a fundamental mechanism for intracellular protein homeostasis in the earliest eukaryotic cells[46].
The comparative analysis of autophagy mechanisms across plants and animals reveals important principles about autophagy evolution[46]. In plants such as Arabidopsis thaliana, mutants defective in ATG genes including atg16 homologs show severe phenotypes including early senescence, starvation sensitivity, and accumulation of protein aggregates, similar to the phenotypes observed in yeast and mammalian systems[46]. This conservation of phenotype despite substantial evolutionary divergence underscores the fundamental importance of the autophagy machinery in eukaryotic cell biology[46]. The identification of autophagy-independent functions of ATG16 in both mammalian and plant systems suggests that these functions may represent conserved, non-redundant roles of ATG16 beyond its canonical autophagy function[46].
Integration into Broader Autophagy Networks
ATG16 functions as a critical hub integrating multiple aspects of autophagy regulation and membrane dynamics. The protein interacts with numerous other autophagy factors beyond those already discussed, including FIP200, which recruits ATG16L1 to sites of phagophore initiation in mammalian cells, and RAB33B, a small GTPase involved in membrane trafficking[13][21][25]. The interaction of ATG16L1 with FIP200 occurs through a distinct binding site (approximately residues 239–246 in ATG16L1) that is spatially adjacent to but separate from the WIPI2 binding site, suggesting that multiple recruitment pathways converge on distinct regions of ATG16L1[13][25]. The interplay between FIP200 and WIPI2 binding to ATG16L1 remains incompletely understood, but evidence suggests that FIP200 binding alone is insufficient to support autophagy, whereas binding to both FIP200 and WIPI2 is required for efficient autophagosome formation[13][25].
The TRIM16 protein has been identified as a scaffold that facilitates autophagy-mediated clearance of protein aggregates through interaction with multiple autophagy factors including ATG16L1[33]. TRIM16 promotes formation of protein aggregates under stress conditions and simultaneously facilitates autophagic degradation of these aggregates by assembling a platform that brings together core autophagic machinery components including ULK1 (autophagy initiation), ATG16L1 (elongation), and LC3B (completion)[33]. This function of TRIM16 illustrates how ATG16L1 participates in broader cellular stress responses beyond the basal autophagy that occurs during nutrient starvation[33].
Conclusion
The atg16 gene in Schizosaccharomyces pombe encodes a highly conserved autophagy protein that functions as a critical structural and catalytic component of the autophagy machinery. ATG16 assembles with ATG12 and ATG5 to form a specialized E3-like ubiquitin ligase that catalyzes the conjugation of the ubiquitin-like protein ATG8 to phosphatidylethanolamine during autophagosome biogenesis. The protein exhibits multiple distinct functional domains: an N-terminal ATG5-binding domain, a central coiled-coil homodimerization domain, and an amphipathic membrane-binding helix. ATG16 localizes to the pre-autophagosomal structure through two partially redundant recruitment mechanisms—a PI3P-dependent pathway mediated by WIPI/Atg18 proteins and a PI3P-independent pathway mediated by interaction with the Atg1 kinase complex. At the PAS, ATG16 functions in membrane tethering and clustering while simultaneously promoting the catalytic transfer of ATG8 to PE, thereby simultaneously organizing membranes and decorating them with lipidated autophagy markers.
Beyond canonical autophagy, emerging evidence indicates that ATG16 participates in autophagy-independent cellular functions including non-canonical autophagy pathways and crosstalk with the ubiquitin proteasome system. The disease association of ATG16L1 variants with Crohn's disease and multiple sclerosis demonstrates the biological significance of this protein in immune homeostasis. The structural features and biochemical mechanisms of ATG16 have been conserved throughout eukaryotic evolution, from single-celled yeasts to mammals, reflecting the fundamental importance of autophagy in cellular homeostasis. Future investigations into ATG16 regulation, its interactions with newly discovered autophagy factors, and its role in disease pathogenesis will likely reveal additional dimensions of ATG16 function and provide insights into the basic mechanisms controlling cellular degradation and recycling pathways.
Citations
- https://www.pombase.org/gene/SPBC405.05
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6356889/
- https://dnaconda.riken.jp/search/SPD_pombe/Sp03/Sp03F02.html
- https://www.uniprot.org/uniprotkb/O94656/entry
- https://www.uniprot.org/uniprotkb/Q676U5/entry
- https://www.cusabio.com/Recombinant-Protein/Recombinant-Schizosaccharomyces-pombe-Meiotic-chromosome-segregation-protein-C4-637693.html
- https://elifesciences.org/articles/70372
- https://pubmed.ncbi.nlm.nih.gov/23064152/
- https://pubmed.ncbi.nlm.nih.gov/24070470/
- https://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=2dym
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3590266/
- https://www.yeastgenome.org/locus/S000004769
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2366851/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC7173070/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC7924733/
- https://elifesciences.org/articles/43088
- https://journals.biologists.com/jcs/article-pdf/133/20/jcs249227/3512148/jcs249227.pdf
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3738441/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5813257/
- https://febs.onlinelibrary.wiley.com/doi/10.1111/febs.15833
- https://www.pombase.org/reference/PMID:23950735
- https://journals.biologists.com/dev/article/144/21/3990/48186/Drosophila-Atg16-promotes-enteroendocrine-cell
- https://pmc.ncbi.nlm.nih.gov/articles/PMC4502675/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2366860/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3877172/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2529362/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC9894987/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3427254/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6138442/
- https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0076237
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5415757/
- http://reactome.org/content/detail/R-HSA-5676229
- https://journals.plos.org/plosgenetics/article?id=10.1371%2Fjournal.pgen.1003715
- https://pmc.ncbi.nlm.nih.gov/articles/PMC4846508/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC9620852/
- https://rupress.org/jcb/article/182/1/129/45336/Quantitative-analysis-of-autophagy-related-protein
- https://www.yeastgenome.org/locus/ATG12
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3288023/
- https://elifesciences.org/articles/39982
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6899017/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8615172/
- https://academic.oup.com/ecco-jcc/article/15/Supplement_1/S155/6286095
- https://www.molbiolcell.org/doi/full/10.1091/mbc.E03-09-0704?view=long&pmid=14699058
- https://dc.uthsc.edu/dissertations/514/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC7265318/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC4111135/
- https://journals.biologists.com/jcs/article/128/2/193/55240/Membrane-dynamics-in-autophagosome-biogenesis
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6405243/
- https://pubmed.ncbi.nlm.nih.gov/25484072/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6028034/
- https://www.bmbreports.org/journal/download_pdf.php?spage=129&volume=48&number=3
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6243101/