Deep Research
Falcon
(IL36RN-deep-research-falcon.md)
Falcon
(IL36RN-deep-research-falcon.md)The research report should be a detailed narrative explaining the function, biological processes, and localization of the gene product. Citations should be given for all claims.
You should prioritize authoritative reviews and primary scientific literature when conducting research. You can supplement
this with annotations you find in gene/protein databases, but these can be outdated or inaccurate.
We are specifically interested in the primary function of the gene - for enzymes, what reaction is catalyzed, and what is the substrate specificity? For transporters, what is the substrate? For structural proteins or adapters, what is the broader structural role? For signaling molecules, what is the role in the pathway.
We are interested in where in or outside the cell the gene product carries out its function.
We are also interested in the signaling or biochemical pathways in which the gene functions. We are less interested in broad pleiotropic effects, except where these elucidate the precise role.
Include evidence where possible. We are interested in both experimental evidence as well as inference from structure, evolution, or bioinformatic analysis. Precise studies should be prioritized over high-throughput, where available.
Research Report: Human IL36RN (UniProt Q9UBH0) — Functional Annotation of Interleukin‑36 Receptor Antagonist (IL‑36Ra)
0. Target verification (mandatory)
The requested target (IL36RN, UniProt Q9UBH0) corresponds to interleukin‑36 receptor antagonist (IL‑36Ra), a member of the IL‑1 cytokine superfamily that antagonizes IL‑36 receptor (IL‑36R) signaling; this mapping is explicit in multiple peer‑reviewed sources focused on IL‑36 biology and generalized pustular psoriasis (GPP). (krueger2024targetedreviewof pages 1-2, hawkes2023theroleof pages 1-3, macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
1. Key concepts and definitions (current understanding)
1.1 What IL36RN encodes
IL36RN encodes IL‑36Ra, an endogenous antagonist within the IL‑36 cytokine subfamily that suppresses pro‑inflammatory responses at epithelial barrier sites, especially skin. (krueger2024targetedreviewof pages 1-2, fukaura2023targetingil36in pages 1-2)
1.2 IL‑36 signaling axis and where IL‑36Ra fits
IL‑36 agonists (IL‑36α/β/γ) signal through IL‑36R with recruitment of the accessory protein IL‑1RAcP, triggering downstream inflammatory pathways including MyD88, NF‑κB, and MAPK activation; IL‑36Ra binds the IL‑36 receptor complex but does not support productive accessory‑protein recruitment, thereby competitively inhibiting IL‑36 signaling. (macleod2016neutrophilelastasemediatedproteolysis pages 1-2, fukaura2023targetingil36in pages 1-2)
A 2023 review further summarizes that IL‑36Ra suppresses signaling by preventing IL‑36R/IL‑1RAcP dimerization and notes canonical downstream inflammatory signaling nodes (MAPK and NF‑κB). (li2023newinsightson pages 1-2)
1.3 Functional definition (primary molecular function)
Primary function: IL‑36Ra is a secreted/extracellular receptor antagonist that inhibits IL‑36R signaling and thereby attenuates IL‑36–driven inflammatory programs (e.g., chemokine induction that recruits neutrophils). (macleod2016neutrophilelastasemediatedproteolysis pages 1-2, li2023newinsightson pages 1-2)
2. Mechanism, regulation, and localization/expression
2.1 Proteolytic processing is required for full activity
A central regulatory concept in IL‑36 biology is protease‑dependent “licensing”: IL‑36 subfamily members are produced as precursors and require N‑terminal processing for full activity (agonist or antagonist). (fukaura2023targetingil36in pages 1-2, macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
IL‑36Ra processing: A key primary study demonstrated that neutrophil elastase (but not other tested neutrophil proteases) cleaves IL‑36Ra into a highly active antagonistic form and that this processed IL‑36Ra more strongly suppresses IL‑36γ‑induced chemokines. (macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
Experimental evidence: In human primary dermal fibroblasts, keratinocytes, and skin equivalents, elastase‑processed IL‑36Ra caused a dose‑dependent reduction of IL‑36γ‑induced IL‑8 and CCL20, supporting the model that proteolytic activation of IL‑36Ra can counter‑regulate neutrophilic inflammation. (macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
Broader protease landscape (IL‑36 family): A 2023 skin‑focused review summarizes that IL‑36 precursor activation can be mediated by neutrophil proteases (cathepsin G, proteinase 3, elastase) and by cathepsin S released from keratinocytes/fibroblasts, and highlights endogenous protease inhibitors (SERPINA1/SERPINA3) as regulators of IL‑36 processing. (fukaura2023targetingil36in pages 1-2)
2.2 Tissue/cellular context and functional localization
IL‑36 biology is concentrated at barrier tissues (skin prominently; also lung and gut). (li2023newinsightson pages 1-2)
In skin, IL‑36 agonists are mainly produced by keratinocytes in the epidermis, with additional production by dendritic cells, macrophages, endothelial cells, and dermal fibroblasts; IL‑36Ra functions at the receptor complex in the extracellular compartment where these cytokines act. (fukaura2023targetingil36in pages 1-2, macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
3. Disease associations with mechanistic clarity (IL36RN loss of function)
3.1 DITRA and generalized pustular psoriasis (GPP)
Loss‑of‑function IL36RN variants cause deficiency of interleukin‑36 receptor antagonist (DITRA) and are strongly linked to severe pustular phenotypes including GPP, consistent with a mechanism of unopposed IL‑36 signaling and exaggerated neutrophilic inflammation. (okorie2024cutaneousfindingsand pages 11-12, macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
3.2 Genetics and mutation frequencies (recent synthesis)
A 2024 targeted review/meta‑analysis reported that IL36RN mutations are significantly more frequent in “GPP‑only” than in GPP with concomitant plaque psoriasis (OR 3.51; 95% CI 2.29–5.38). (krueger2024targetedreviewof pages 1-2)
In that synthesis, monoallelic IL36RN variants were reported in up to 33.3% of GPP patients and biallelic variants in up to 73.2%, compared with monoallelic 0%–11.9% and biallelic 0% in plaque psoriasis only. (krueger2024targetedreviewof pages 1-2)
Frequently reported variants included c.115+6T>C (p.Arg10ArgfsX1), c.227C>T (p.Pro76Leu), and c.338C>T (p.Ser113Leu), with geographic/ethnic variation (notably higher prevalence of the most frequent mutation in East Asian and Asian‑enriched studies). (krueger2024targetedreviewof pages 1-2)
4. Recent developments (2023–2024 prioritized)
4.1 Consolidation of IL‑36 as a therapeutic axis in inflammatory skin disease
A 2023 BioDrugs review summarizes IL‑36 cytokines as key regulators of innate and adaptive immunity in skin and highlights that anti‑IL‑36 agents (notably IL‑36R‑blocking antibodies) have been evaluated across multiple inflammatory dermatoses. (fukaura2023targetingil36in pages 1-2)
4.2 Updated clinical framing of GPP severity and epidemiology
A 2023 Frontiers in Immunology review emphasizes that GPP is a rare, potentially life‑threatening inflammatory disease driven by abnormal activation of the IL‑36–chemokine–neutrophil axis, and reports prevalence estimates ranging approximately 2–120 cases per million. (hawkes2023theroleof pages 1-3)
The same review summarizes mortality estimates reported across studies (e.g., 0–3.3 deaths per 100 patient‑years) and notes a Japanese hospitalized cohort (N=1516) reporting 4.2% mortality. (hawkes2023theroleof pages 1-3)
4.3 Genetics-to-therapy linkage strengthened by 2024 mutation meta-analysis
The 2024 meta-analytic synthesis (above) provides a clearer basis for using IL36RN genotype to distinguish GPP subgroups and to motivate pathway‑targeted approaches. (krueger2024targetedreviewof pages 1-2)
5. Current applications and real‑world implementations (therapeutics)
5.1 IL‑36R blockade with spesolimab (approved therapy)
Regulatory status: Spesolimab (anti‑IL‑36R monoclonal antibody) received US FDA approval in September 2022 for treatment of GPP flares in adults, with subsequent approvals in other regions; a 2024 review also notes more recent approval for subcutaneous dosing for treatment when not in flare (per label). (gwillim2024spesolimabforgeneralized pages 2-4)
Dosing for flares (as summarized in 2024 review): single 900 mg IV over 90 minutes, with an optional second 900 mg dose 1 week later if symptoms persist. (gwillim2024spesolimabforgeneralized pages 2-4)
Evidence: rapid flare control (key statistics)
A 2024 review summarizing the two trials supporting initial approval reported:
-
Phase 1 proof‑of‑concept (NCT02978690; n=7): 5/7 (71%) achieved GPPGA 0/1 by week 1 and 7/7 by week 4; mean GPPASI improvement was 59.0% at week 1, 73.2% at week 2, and 79.8% at week 4. (gwillim2024spesolimabforgeneralized pages 1-2, gwillim2024spesolimabforgeneralized pages 4-5)
-
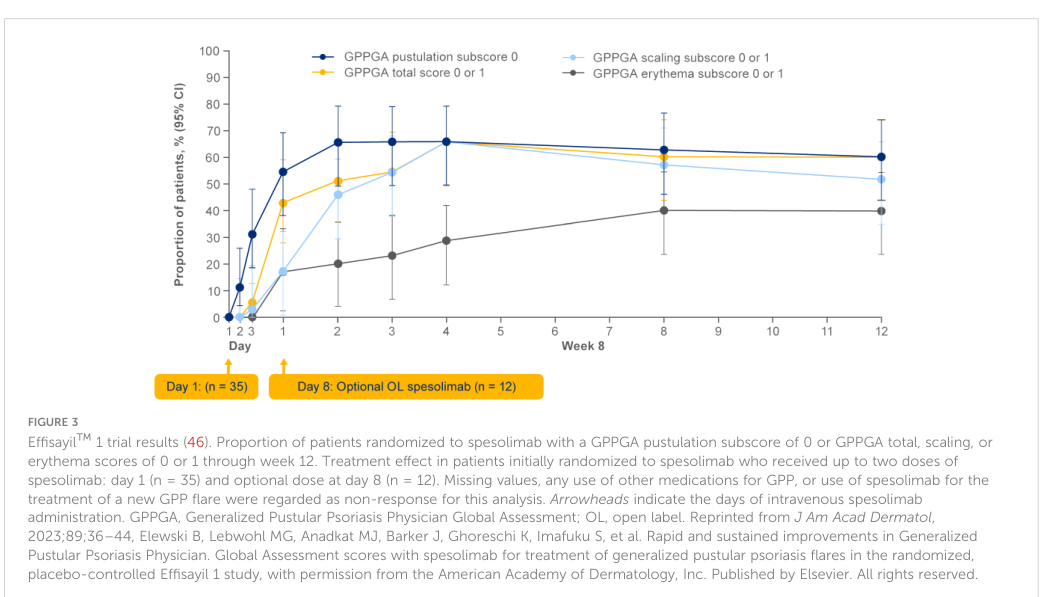
Effisayil™1 (NCT03782792; phase 2, randomized): At week 1, GPPGA pustulation subscore 0 occurred in 19/35 (54%) with spesolimab vs 1/18 (6%) placebo (difference 49 percentage points; 95% CI 21–67; P<0.001). GPPGA total score 0/1 occurred in 15/35 (43%) vs 2/18 (11%) (difference 32 percentage points; 95% CI 2–53; P=0.02). (gwillim2024spesolimabforgeneralized pages 1-2, gwillim2024spesolimabforgeneralized media 67b8352d)
Safety statistic (week 1): infections were reported in 6/35 (17%) spesolimab vs 1/18 (6%) placebo. (gwillim2024spesolimabforgeneralized pages 1-2, gwillim2024spesolimabforgeneralized pages 5-6)
Evidence: flare prevention/maintenance dosing
A 2024 Pharmaceutics review summarized Effisayil™2 (dose‑finding, 48 weeks; n=123), reporting flare occurrence by week 48 of 52% (placebo), 23% (low dose), 29% (medium dose), and 10% (high dose), with statistically significant superiority for time‑to‑flare (reported p=0.0005). (vilaca2024newandemerging pages 6-8, vilaca2024newandemerging pages 5-6)
5.2 Other IL‑36R inhibitors in clinical development: imsidolimab (ANB019)
A 2024 Pharmaceutics review summarized an open‑label phase 2 single‑arm study (GALLOP; n=8) reporting 75% clinical response by Clinical Global Impression at weeks 4 and 16, with 50% described as “very much improved.” (vilaca2024newandemerging pages 6-8)
6. Expert opinions and analysis (authoritative synthesis)
6.1 Consensus mechanistic framing: “IL‑36–chemokine–neutrophil axis”
A 2023 Frontiers in Immunology review explicitly frames GPP as driven by abnormal activation of the IL‑36–chemokine–neutrophil axis, providing a mechanistic justification for IL‑36R antagonism as a targeted therapy strategy. (hawkes2023theroleof pages 1-3)
6.2 Therapeutic rationale: rebalancing agonist vs antagonist activity
Skin‑focused reviews emphasize that IL‑36 biology functions as a regulated system in which excessive agonist activity, reduced antagonist function (including IL36RN deficiency), and protease‑mediated activation together determine inflammatory outcomes—supporting two therapeutic concepts: (i) direct IL‑36R blockade (e.g., spesolimab) and (ii) upstream modulation of protease processing (e.g., elastase/cathepsin pathways). (fukaura2023targetingil36in pages 1-2, macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
7. Key statistics and data (from recent studies)
7.1 IL36RN mutation frequencies in GPP (2024 synthesis)
- IL36RN mutation enrichment in GPP‑only vs GPP+plaque psoriasis: OR 3.51 (95% CI 2.29–5.38). (krueger2024targetedreviewof pages 1-2)
- GPP: monoallelic variants up to 33.3%, biallelic up to 73.2% (vs plaque psoriasis only: monoallelic 0–11.9%, biallelic 0%). (krueger2024targetedreviewof pages 1-2)
7.2 Spesolimab efficacy in acute flares (key endpoints)
- Effisayil 1 week‑1 pustulation clearance: 54% vs 6% placebo (P<0.001). (gwillim2024spesolimabforgeneralized pages 1-2, gwillim2024spesolimabforgeneralized media 67b8352d)
- Effisayil 1 week‑1 “clear/almost clear” (GPPGA 0/1): 43% vs 11% placebo (P=0.02). (gwillim2024spesolimabforgeneralized pages 1-2, gwillim2024spesolimabforgeneralized media 67b8352d)
- Week‑1 infections: 17% vs 6% placebo. (gwillim2024spesolimabforgeneralized pages 1-2, gwillim2024spesolimabforgeneralized pages 5-6)
7.3 GPP epidemiology and severity
- Prevalence estimates: ~2–120 cases per million (region‑dependent). (hawkes2023theroleof pages 1-3)
- Mortality: variable across studies; example estimates include 0–3.3 deaths per 100 patient‑years and 4.2% mortality in a Japanese hospitalized cohort (N=1516). (hawkes2023theroleof pages 1-3)
Evidence map (compact)
The following table consolidates the functional annotation and translational evidence for IL36RN/IL‑36Ra, emphasizing 2023–2024 sources.
| Aspect | Key points (1-2 sentences) | Representative evidence (include what was measured/observed) | Key recent sources (2023-2024 prioritized) with URL and publication month/year | Context IDs |
|---|---|---|---|---|
| Identity/definition | IL36RN encodes interleukin-36 receptor antagonist (IL-36Ra), the natural antagonist of the IL-36 pathway in humans; this matches UniProt Q9UBH0 and the IL-1 family context. In GPP-focused reviews, IL-36Ra is described as a suppressor of proinflammatory responses whose insufficiency permits neutrophil-dominant sterile pustular inflammation. | Reviews describe IL36RN as the gene encoding IL-36Ra and note that dysregulated IL-36 signaling drives neutrophil infiltration and pustule formation in generalized pustular psoriasis (GPP). | Krueger et al., Skin Health and Disease (Mar 2024), https://doi.org/10.1002/ski2.343; Hawkes et al., Frontiers in Immunology (Nov 2023), https://doi.org/10.3389/fimmu.2023.1292941 | (krueger2024targetedreviewof pages 1-2, hawkes2023theroleof pages 1-3) |
| Molecular mechanism of antagonism | IL-36Ra binds the same receptor axis as IL-36 agonists but prevents productive signaling; mechanistically, it blocks formation of the signaling-competent IL-36R/IL-1RAcP complex and thereby suppresses downstream MyD88–NF-κB/MAPK activation. This is the primary molecular function of IL36RN. | Experimental and review evidence indicates IL-36Ra binds IL-36R without accessory-protein recruitment; downstream inflammatory outputs such as chemokines/cytokines are consequently reduced. One review additionally notes higher-affinity/slower-off-rate receptor binding than agonists and prevention of receptor dimerization. | Fukaura & Akiyama, BioDrugs (Mar 2023), https://doi.org/10.1007/s40259-023-00587-5; Li et al., Experimental and Therapeutic Medicine (Apr 2023), https://doi.org/10.3892/etm.2023.11974 | (fukaura2023targetingil36in pages 1-2, li2023newinsightson pages 1-2) |
| Proteolytic processing/activating proteases | Like other IL-1 family cytokines, IL-36Ra requires N-terminal processing for full antagonistic activity. For IL-36Ra specifically, neutrophil elastase cleaves the precursor into a highly active antagonistic form; by contrast, IL-36 agonists are activated by proteases including cathepsin G, proteinase 3, elastase, and cathepsin S. | In primary human dermal fibroblasts, keratinocytes, and skin equivalents, cleaved IL-36Ra reduced IL-36γ-induced IL-8 and CCL20 more effectively than full-length IL-36Ra; the 2016 study identified elastase, but not other tested neutrophil proteases, as the activating protease for IL-36Ra. Reviews summarize broader IL-36-family protease control and SERPINA1/SERPINA3 inhibition of elastase/cathepsin G. | Macleod et al., Scientific Reports (Apr 2016), https://doi.org/10.1038/srep24880; Fukaura & Akiyama, BioDrugs (Mar 2023), https://doi.org/10.1007/s40259-023-00587-5 | (macleod2016neutrophilelastasemediatedproteolysis pages 1-2, fukaura2023targetingil36in pages 1-2) |
| Expression/localization (skin/barrier tissues, cell types) | IL-36 biology is centered at barrier tissues. Recent reviews place IL-36 ligands/receptor broadly in skin, lung, and intestine, with skin-relevant production from keratinocytes, dendritic cells, macrophages, endothelial cells, and dermal fibroblasts; IL-36Ra functions extracellularly at the receptor complex in these barrier environments. | Reviews report that IL-36α/γ are mainly expressed by keratinocytes in epidermis, with additional production by dendritic cells, macrophages, endothelial cells, and dermal fibroblasts; IL-36 ligands and IL-36R are broadly expressed at mucosal/barrier sites. In COPD, airway studies found increased IL-36γ in epithelial-derived compartments and decreased IL-36Ra in bronchoalveolar/nasal fluid, supporting extracellular pathway imbalance. | Fukaura & Akiyama, BioDrugs (Mar 2023), https://doi.org/10.1007/s40259-023-00587-5; Li et al., Experimental and Therapeutic Medicine (Apr 2023), https://doi.org/10.3892/etm.2023.11974 | (fukaura2023targetingil36in pages 1-2, li2023newinsightson pages 1-2) |
| Disease genetics (DITRA/GPP; mutation frequencies and notable variants) | Loss-of-function IL36RN variants cause DITRA and are strongly enriched in GPP, especially GPP without plaque psoriasis. Recent meta-analytic review found monoallelic variants in up to 33.3% and biallelic variants in up to 73.2% of GPP patients, versus 0%-11.9% monoallelic and 0% biallelic in plaque psoriasis only; common variants include c.115+6T>C (p.Arg10ArgfsX1), c.227C>T (p.Pro76Leu), and c.338C>T (p.Ser113Leu). | Meta-analysis reported a significantly higher IL36RN mutation rate in GPP-only vs GPP+plaque psoriasis (OR 3.51, 95% CI 2.29-5.38). Case-based DITRA review documents pathogenic homozygous variants and notes that IL-36 ligands and IL-36Ra require proteolytic processing for full activity. | Krueger et al., Skin Health and Disease (Mar 2024), https://doi.org/10.1002/ski2.343; Okorie et al., Experimental Dermatology (Sep 2024), https://doi.org/10.1111/exd.14934 | (krueger2024targetedreviewof pages 1-2, okorie2024cutaneousfindingsand pages 11-12) |
| Therapeutic targeting (spesolimab approvals; Effisayil 1 and phase 1 response rates; Effisayil 2 flare prevention; imsidolimab early data) | Spesolimab is a first-in-class anti-IL-36R monoclonal antibody approved first by the US FDA in Sep 2022 for adult GPP flares, with later approvals in other regions and more recent subcutaneous maintenance approval. Clinical efficacy is rapid in acute flares and promising for flare prevention; imsidolimab (ANB019) has shown early activity but less mature evidence. | Phase 1 proof-of-concept: 5/7 (71%) achieved GPPGA 0/1 by week 1, 7/7 by week 4; mean GPPASI improved 59.0% (wk1), 73.2% (wk2), 79.8% (wk4), with no severe/serious AEs reported. Effisayil 1: week-1 GPPGA pustulation 0 in 19/35 (54%) on spesolimab vs 1/18 (6%) placebo (difference 49 percentage points; 95% CI 21-67; P<0.001); GPPGA total 0/1 in 15/35 (43%) vs 2/18 (11%) (difference 32 points; 95% CI 2-53; P=0.02); infections at week 1 in 17% vs 6%. Effisayil 2: over 48 weeks, flares occurred in 52% placebo, 23% low-dose, 29% medium-dose, and 10% high-dose spesolimab; time-to-flare superiority reported (p=0.0005). Imsidolimab: open-label GALLOP study (n=8) reported 75% clinical response at weeks 4 and 16, with 50% rated “very much improved.” | Gwillim & Nichols, Frontiers in Immunology (Jul 2024), https://doi.org/10.3389/fimmu.2024.1359481; Vilaça et al., Pharmaceutics (Jul 2024), https://doi.org/10.3390/pharmaceutics16070908 | (gwillim2024spesolimabforgeneralized pages 1-2, gwillim2024spesolimabforgeneralized pages 5-6, vilaca2024newandemerging pages 5-6, vilaca2024newandemerging pages 6-8, gwillim2024spesolimabforgeneralized pages 2-4, gwillim2024spesolimabforgeneralized media 67b8352d) |
| Epidemiology/mortality stats for GPP | GPP is rare but clinically serious, and IL36RN biology is most clearly translated in this disease context. Recent reviews estimate prevalence at roughly 2-120 cases per million, with reported mortality ranging from 0-3.3 deaths per 100 patient-years in some studies; a Japanese hospitalized cohort (N=1516) reported 4.2% mortality. | Review text summarizes regional prevalence variability and mortality, and emphasizes that severe flares may require emergency or inpatient care because of complications such as sepsis, heart failure, renal failure, and death. | Hawkes et al., Frontiers in Immunology (Nov 2023), https://doi.org/10.3389/fimmu.2023.1292941; Gwillim & Nichols, Frontiers in Immunology (Jul 2024), https://doi.org/10.3389/fimmu.2024.1359481 | (hawkes2023theroleof pages 1-3, gwillim2024spesolimabforgeneralized pages 2-4) |
Table: This table summarizes verified human IL36RN/IL-36Ra biology, disease genetics, and translational evidence, emphasizing 2023-2024 reviews and clinical data. It is useful as a compact evidence map connecting core function to therapeutics such as spesolimab.
References (URLs and publication dates)
- Fukaura R, Akiyama M. Targeting IL‑36 in Inflammatory Skin Diseases. BioDrugs. Published online 3 Mar 2023. https://doi.org/10.1007/s40259-023-00587-5 (fukaura2023targetingil36in pages 1-2)
- Li M, Jiang W, Wang Z, et al. New insights on IL‑36 in intestinal inflammation and colorectal cancer (Review). Experimental and Therapeutic Medicine. Apr 2023. https://doi.org/10.3892/etm.2023.11974 (li2023newinsightson pages 1-2)
- Hawkes JE, Visvanathan S, Krueger JG. The role of the interleukin‑36 axis in generalized pustular psoriasis: a review of the mechanism of action of spesolimab. Frontiers in Immunology. Published 21 Nov 2023. https://doi.org/10.3389/fimmu.2023.1292941 (hawkes2023theroleof pages 1-3)
- Krueger JG, Pagotto A, Haftel S, Gradl B. Targeted review of IL36RN mutations in patients with generalised pustular psoriasis. Skin Health and Disease. Mar 2024. https://doi.org/10.1002/ski2.343 (krueger2024targetedreviewof pages 1-2)
- Okorie CL, Nayudu K, Nambudiri VE. Cutaneous findings and treatments in deficiency of interleukin‑36 receptor antagonist (DITRA): A review of the literature. Experimental Dermatology. Sep 2024. https://doi.org/10.1111/exd.14934 (okorie2024cutaneousfindingsand pages 11-12)
- Gwillim EC, Nichols AJ. Spesolimab for generalized pustular psoriasis: a review of two key clinical trials supporting initial US regulatory approval. Frontiers in Immunology. Jul 2024. https://doi.org/10.3389/fimmu.2024.1359481 (gwillim2024spesolimabforgeneralized pages 1-2)
- Vilaça J, Yilmaz O, Torres T. New and Emerging Treatments for Generalized Pustular Psoriasis: Focus on IL‑36 Receptor Inhibitors. Pharmaceutics. Jul 2024. https://doi.org/10.3390/pharmaceutics16070908 (vilaca2024newandemerging pages 5-6)
- Macleod T, Doble R, McGonagle D, et al. Neutrophil elastase‑mediated proteolysis activates the anti‑inflammatory cytokine IL‑36 receptor antagonist. Scientific Reports. Published 22 Apr 2016. https://doi.org/10.1038/srep24880 (macleod2016neutrophilelastasemediatedproteolysis pages 1-2)
References
-
(krueger2024targetedreviewof pages 1-2): James G. Krueger, Anna Pagotto, Samuel Haftel, and Birgit Gradl. Targeted review of il36rn mutations in patients with generalised pustular psoriasis. Skin Health and Disease, Mar 2024. URL: https://doi.org/10.1002/ski2.343, doi:10.1002/ski2.343. This article has 13 citations and is from a peer-reviewed journal.
-
(hawkes2023theroleof pages 1-3): Jason E. Hawkes, Sudha Visvanathan, and James G. Krueger. The role of the interleukin-36 axis in generalized pustular psoriasis: a review of the mechanism of action of spesolimab. Frontiers in Immunology, Nov 2023. URL: https://doi.org/10.3389/fimmu.2023.1292941, doi:10.3389/fimmu.2023.1292941. This article has 32 citations and is from a peer-reviewed journal.
-
(macleod2016neutrophilelastasemediatedproteolysis pages 1-2): Tom Macleod, Rosella Doble, Dennis McGonagle, Christopher W. Wasson, Adewonuola Alase, Martin Stacey, and Miriam Wittmann. Neutrophil elastase-mediated proteolysis activates the anti-inflammatory cytokine il-36 receptor antagonist. Scientific Reports, Apr 2016. URL: https://doi.org/10.1038/srep24880, doi:10.1038/srep24880. This article has 120 citations and is from a peer-reviewed journal.
-
(fukaura2023targetingil36in pages 1-2): Ryo Fukaura and Masashi Akiyama. Targeting il-36 in inflammatory skin diseases. BioDrugs, pages 1-15, Mar 2023. URL: https://doi.org/10.1007/s40259-023-00587-5, doi:10.1007/s40259-023-00587-5. This article has 44 citations and is from a peer-reviewed journal.
-
(li2023newinsightson pages 1-2): Minghui Li, Wei Jiang, Zehui Wang, Yihan Lu, and Jun Zhang. New insights on il‑36 in intestinal inflammation and colorectal cancer (review). Experimental and Therapeutic Medicine, Apr 2023. URL: https://doi.org/10.3892/etm.2023.11974, doi:10.3892/etm.2023.11974. This article has 10 citations and is from a peer-reviewed journal.
-
(okorie2024cutaneousfindingsand pages 11-12): Chiamaka L. Okorie, Krithika Nayudu, and Vinod E. Nambudiri. Cutaneous findings and treatments in deficiency of interleukin‐36 receptor antagonist (ditra): a review of the literature. Experimental Dermatology, Sep 2024. URL: https://doi.org/10.1111/exd.14934, doi:10.1111/exd.14934. This article has 15 citations and is from a domain leading peer-reviewed journal.
-
(gwillim2024spesolimabforgeneralized pages 2-4): Eran C. Gwillim and Anna J. Nichols. Spesolimab for generalized pustular psoriasis: a review of two key clinical trials supporting initial us regulatory approval. Frontiers in Immunology, Jul 2024. URL: https://doi.org/10.3389/fimmu.2024.1359481, doi:10.3389/fimmu.2024.1359481. This article has 17 citations and is from a peer-reviewed journal.
-
(gwillim2024spesolimabforgeneralized pages 1-2): Eran C. Gwillim and Anna J. Nichols. Spesolimab for generalized pustular psoriasis: a review of two key clinical trials supporting initial us regulatory approval. Frontiers in Immunology, Jul 2024. URL: https://doi.org/10.3389/fimmu.2024.1359481, doi:10.3389/fimmu.2024.1359481. This article has 17 citations and is from a peer-reviewed journal.
-
(gwillim2024spesolimabforgeneralized pages 4-5): Eran C. Gwillim and Anna J. Nichols. Spesolimab for generalized pustular psoriasis: a review of two key clinical trials supporting initial us regulatory approval. Frontiers in Immunology, Jul 2024. URL: https://doi.org/10.3389/fimmu.2024.1359481, doi:10.3389/fimmu.2024.1359481. This article has 17 citations and is from a peer-reviewed journal.
-
(gwillim2024spesolimabforgeneralized media 67b8352d): Eran C. Gwillim and Anna J. Nichols. Spesolimab for generalized pustular psoriasis: a review of two key clinical trials supporting initial us regulatory approval. Frontiers in Immunology, Jul 2024. URL: https://doi.org/10.3389/fimmu.2024.1359481, doi:10.3389/fimmu.2024.1359481. This article has 17 citations and is from a peer-reviewed journal.
-
(gwillim2024spesolimabforgeneralized pages 5-6): Eran C. Gwillim and Anna J. Nichols. Spesolimab for generalized pustular psoriasis: a review of two key clinical trials supporting initial us regulatory approval. Frontiers in Immunology, Jul 2024. URL: https://doi.org/10.3389/fimmu.2024.1359481, doi:10.3389/fimmu.2024.1359481. This article has 17 citations and is from a peer-reviewed journal.
-
(vilaca2024newandemerging pages 6-8): João Vilaça, Orhan Yilmaz, and Tiago Torres. New and emerging treatments for generalized pustular psoriasis: focus on il-36 receptor inhibitors. Pharmaceutics, 16:908, Jul 2024. URL: https://doi.org/10.3390/pharmaceutics16070908, doi:10.3390/pharmaceutics16070908. This article has 13 citations.
-
(vilaca2024newandemerging pages 5-6): João Vilaça, Orhan Yilmaz, and Tiago Torres. New and emerging treatments for generalized pustular psoriasis: focus on il-36 receptor inhibitors. Pharmaceutics, 16:908, Jul 2024. URL: https://doi.org/10.3390/pharmaceutics16070908, doi:10.3390/pharmaceutics16070908. This article has 13 citations.
Artifacts
Citations
- li2023newinsightson pages 1-2
- macleod2016neutrophilelastasemediatedproteolysis pages 1-2
- krueger2024targetedreviewof pages 1-2
- hawkes2023theroleof pages 1-3
- gwillim2024spesolimabforgeneralized pages 2-4
- vilaca2024newandemerging pages 6-8
- okorie2024cutaneousfindingsand pages 11-12
- gwillim2024spesolimabforgeneralized pages 1-2
- vilaca2024newandemerging pages 5-6
- gwillim2024spesolimabforgeneralized pages 4-5
- gwillim2024spesolimabforgeneralized pages 5-6
- https://doi.org/10.1002/ski2.343;
- https://doi.org/10.3389/fimmu.2023.1292941
- https://doi.org/10.1007/s40259-023-00587-5;
- https://doi.org/10.3892/etm.2023.11974
- https://doi.org/10.1038/srep24880;
- https://doi.org/10.1007/s40259-023-00587-5
- https://doi.org/10.1111/exd.14934
- https://doi.org/10.3389/fimmu.2024.1359481;
- https://doi.org/10.3390/pharmaceutics16070908
- https://doi.org/10.3389/fimmu.2023.1292941;
- https://doi.org/10.3389/fimmu.2024.1359481
- https://doi.org/10.1002/ski2.343
- https://doi.org/10.1038/srep24880
- https://doi.org/10.1002/ski2.343,
- https://doi.org/10.3389/fimmu.2023.1292941,
- https://doi.org/10.1038/srep24880,
- https://doi.org/10.1007/s40259-023-00587-5,
- https://doi.org/10.3892/etm.2023.11974,
- https://doi.org/10.1111/exd.14934,
- https://doi.org/10.3389/fimmu.2024.1359481,
- https://doi.org/10.3390/pharmaceutics16070908,
OpenAI
(IL36RN-deep-research-openai.md)
OpenAI
(IL36RN-deep-research-openai.md)Introduction and Gene Overview
IL36RN (Interleukin-36 receptor antagonist) is a human gene (UniProt Q9UBH0) encoding a cytokine of the interleukin-1 (IL-1) family. It is the sole antagonist ligand in the IL-36 subfamily of cytokines, which also includes three pro-inflammatory agonists (IL-36α, IL-36β, and IL-36γ) (www.spandidos-publications.com) (www.spandidos-publications.com). Discovered around 2000 in the IL-1 gene cluster on chromosome 2, IL-36RN is highly homologous to IL-1 cytokines in sequence and predicted structure (www.spandidos-publications.com). The IL-36RN protein is also known by synonyms IL-1F5 or IL-36Ra, reflecting its function as the IL-36 receptor antagonist (www.proteinatlas.org). This protein is a member of the IL-1 family’s β-trefoil fold cytokines, and like other IL-1 family members it is primarily active outside the cell, regulating immune signaling by binding to cell-surface cytokine receptors (academic.oup.com) (academic.oup.com). Notably, IL-36Ra lacks a conventional signal peptide, but it is released into the extracellular space (likely via unconventional secretion or cell damage) where it can exert its function (academic.oup.com). It is produced by a variety of cell types in barrier tissues – for example, keratinocytes in the skin and immune cells (monocytes, macrophages, dendritic cells, etc.) are known to express IL-36Ra, often alongside the IL-36 agonists (academic.oup.com) (academic.oup.com). In resting conditions, certain myeloid cells even express IL-36Ra constitutively, suggesting a basal level of antagonist is maintained to prevent spontaneous inflammation (academic.oup.com). Overall, IL36RN encodes a cytokine that serves as a key negative regulator in the IL-36 signaling axis, helping maintain immune homeostasis at epithelial barriers.
Molecular Function: IL-36 Receptor Antagonist Activity
The IL-36RN gene product (IL-36Ra protein) functions as a competitive antagonist of IL-36 cytokine signaling. It binds to the IL-36 receptor (IL-36R, also known as IL1RL2 or IL-1Rrp2) on the cell surface with high affinity, but – in contrast to the IL-36 agonists – IL-36Ra does not recruit the signaling co-receptor required for downstream activation (academic.oup.com). Specifically, IL-36 agonist ligands normally engage a receptor complex of IL-36R and the IL-1 receptor accessory protein (IL-1RAcP), which brings together intracellular TIR domains and triggers MyD88-dependent signaling cascades (activating NF-κB and MAP kinases) (pmc.ncbi.nlm.nih.gov) (www.spandidos-publications.com). IL-36Ra can bind to IL-36R in place of the agonist, but its binding prevents IL-1RAcP recruitment and receptor dimerization, thereby blocking the formation of a functional signaling complex (www.spandidos-publications.com). In essence, IL-36Ra occupies the receptor without triggering it – earning its designation as a “true antagonist” of IL-36 signaling (academic.oup.com). This mechanism halts the downstream biochemical pathway: when IL-36Ra is bound, the IL-36R/IL-1RAcP complex cannot assemble, and thus the typical IL-36-induced activation of NF-κB, MAPK, and pro-inflammatory gene expression is suppressed (www.spandidos-publications.com).
Importantly, IL-36Ra’s binding affinity for the IL-36 receptor is substantially stronger than that of the agonist cytokines, which enables effective competitive inhibition. A biochemical kinetics study (J. Biol. Chem. 2017) measured IL-36Ra’s binding and showed a dissociation constant (K_D) of roughly 5–6 nM for IL-36R, compared to much weaker affinities of ~480 nM for IL-36α and ~1800 nM for IL-36γ (pmc.ncbi.nlm.nih.gov). In the same analysis, IL-36Ra was found to dissociate from the receptor extremely slowly (a markedly slower off-rate than the agonists) (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). This means once IL-36Ra is bound to IL-36R, it tends to remain bound, effectively outcompeting IL-36α/β/γ for receptor occupancy. These quantitative binding data provide a clear mechanistic basis for IL-36Ra’s function: by tightly and persistently occupying IL-36R, IL-36Ra prevents the pro-inflammatory IL-36 cytokines from delivering their signal (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). In summary, the primary biochemical function of the IL36RN gene product is to serve as a high-affinity receptor “blocker” that safeguards cells from excessive IL-36–mediated immunity. This antagonist action is conceptually analogous to the IL-1 receptor antagonist (IL-1Ra) in the IL-1 system, underlining a broader theme in IL-1 family biology where dedicated anti-cytokines modulate the activity of their pro-inflammatory counterparts (academic.oup.com).
Activation and Processing Requirements
Like many IL-1 family cytokines, IL-36Ra is synthesized as a precursor that requires proteolytic processing to attain full activity. The unprocessed IL-36Ra protein (155 amino acids in length) has an N-terminal extension that hinders its receptor-binding capacity until removed (pubmed.ncbi.nlm.nih.gov). In vitro studies first showed that removing the very N-terminal methionine (and adjacent residues) dramatically increases IL-36Ra’s antagonist potency (pubmed.ncbi.nlm.nih.gov). This is analogous to IL-36 agonists, which also require N-terminal truncation by proteases (such as neutrophil proteases) to become fully active cytokines (www.spandidos-publications.com) (www.spandidos-publications.com). For IL-36Ra, neutrophil-derived proteases have been identified as key activating enzymes: neutrophil elastase can cleave IL-36Ra’s N-terminus, converting it into a significantly more potent antagonist form (www.spandidos-publications.com). Macleod et al. (2016) demonstrated that elastase processing of IL-36Ra increases its ability to block IL-36 signaling, effectively “unlocking” its anti-inflammatory function (www.spandidos-publications.com).
Crucially, there is strong in vivo evidence that N-terminal processing is required for IL-36Ra’s function. A 2017 clinical genetics study reported a naturally occurring IL36RN mutation (p.V2F) in patients with autoinflammatory skin disease which impairs IL-36Ra processing (pubmed.ncbi.nlm.nih.gov). This missense mutation (valine to phenylalanine at position 2) did not prevent IL-36Ra protein expression, but it prevented the removal of the N-terminal methionine. The mutant IL-36Ra retained its initiator Met and was essentially devoid of antagonist activity (pubmed.ncbi.nlm.nih.gov). Mass spectrometry confirmed that the pathogenic IL-36Ra variant was not being properly N-terminally trimmed in vivo (pubmed.ncbi.nlm.nih.gov). These findings provided the first direct evidence in humans that cleavage of the N-terminus is a mandatory step for IL-36Ra to achieve optimal receptor antagonist function (pubmed.ncbi.nlm.nih.gov). Without this maturation step, IL-36Ra cannot effectively bind IL-36R and block signaling, leading to unrestrained IL-36 activity in tissues. In summary, IL-36Ra is an inactive pro-cytokine until proteolytic processing (e.g. removal of the first methionine and likely adjacent residues) generates the active form (pubmed.ncbi.nlm.nih.gov) (www.spandidos-publications.com). This regulatory mechanism ensures that IL-36Ra’s inhibitory activity can be controlled – for instance, activated neutrophils in inflamed tissue can activate IL-36Ra when and where it is needed by cleaving it with elastase (www.spandidos-publications.com). Processing thus serves as a post-translational switch that licenses IL-36RN’s gene product to carry out its immune-modulatory function.
Biological Role in Immune Processes and Pathways
IL-36Ra is a key regulator of inflammatory responses, particularly in skin and other epithelial barriers. By antagonizing IL-36 signaling, it keeps certain innate immune pathways in check to prevent damage from excessive inflammation. In the skin, IL-36 cytokines are potent drivers of keratinocyte and myeloid cell activation, and IL-36Ra provides critical counter-regulation. The importance of IL36RN in cutaneous immunity is highlighted by the rare autoinflammatory disease resulting from its loss. Patients with homozygous loss-of-function mutations in IL36RN develop generalized pustular psoriasis (GPP), a severe episodic inflammatory skin disease (pmc.ncbi.nlm.nih.gov) (academic.oup.com). GPP is characterized by widespread sterile pustules and systemic inflammation, and these cases (collectively termed DITRA – Deficiency of IL-36Ra) revealed the essential role of IL-36Ra in controlling skin inflammation. The discovery in 2011 that IL36RN mutations cause GPP underscored that unopposed IL-36 signaling can provoke life-threatening inflammation, establishing IL-36Ra as a critical “brake” in skin innate immunity (pubmed.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). Indeed, experimental models confirm this: IL-36Ra–deficient mice exhibit exacerbated skin inflammation under challenge conditions (academic.oup.com). In psoriatic skin lesions, IL36RN expression is actually upregulated along with IL-36α/β/γ, suggesting a physiological feedback mechanism — the tissue increases production of the antagonist IL-36Ra in an attempt to counteract the high levels of IL-36 agonists in psoriasis (academic.oup.com).
Within the IL-36 signaling pathway, IL-36Ra’s role is to modulate the activity of a classic pro-inflammatory cascade. When IL-36 agonists bind IL-36R on target cells (such as keratinocytes, dendritic cells, monocytes), the receptor complex recruits MyD88 and triggers downstream kinases (IRAKs, TRAF6), culminating in activation of NF-κB and AP-1 transcription factors (pmc.ncbi.nlm.nih.gov). This leads to production of numerous inflammatory mediators – for example, IL-36 signaling can induce chemokines like CXCL1 (KC), IL-8, and cytokines such as IL-6, IL-12, IL-23, thereby amplifying immune cell recruitment and Th17-type responses (pmc.ncbi.nlm.nih.gov) (academic.oup.com). By blocking IL-36R, IL-36Ra prevents these downstream signals, thereby limiting the release of neutrophil-recruiting chemokines and other inflammatory cytokines (pmc.ncbi.nlm.nih.gov). In essence, IL-36Ra helps maintain a balance: it allows initial inflammatory signals needed for host defense or tissue repair but prevents those signals from overshooting. For instance, wound healing studies illustrate this balance – IL36RN knockout mice show delayed wound closure due to excessive, prolonged inflammation. In the absence of IL-36Ra, wounds accumulate abnormally high numbers of neutrophils and macrophages and elevated inflammatory cytokines, impairing normal healing (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). This phenotype was rescued by interventions that block IL-36 signaling or related innate pathways, confirming that the excess inflammation from unchecked IL-36 was the cause of the impaired healing (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). Thus, IL-36Ra’s regulatory role is crucial in the early stages of tissue injury response – it dampens the innate immune cell infiltration and cytokine surge to promote proper resolution and tissue recovery (pmc.ncbi.nlm.nih.gov).
Beyond the skin, IL36RN is increasingly recognized as an important player in other organ systems’ immunity. The IL-36/IL-36Ra axis is active in the gut mucosa, lungs, and joints, where it similarly governs local inflammatory processes (www.spandidos-publications.com) (academic.oup.com). In the intestines, recent research has identified IL-36 signaling as a “key regulator” of intestinal homeostasis and inflammation (pubmed.ncbi.nlm.nih.gov). IL-36 agonists are broadly expressed in inflammatory bowel disease (IBD) lesions, and IL-36RN (IL-36Ra) is thought to counterbalance those signals to maintain mucosal integrity (academic.oup.com) (pubmed.ncbi.nlm.nih.gov). Notably, rare IL36RN mutations have been reported in severe Crohn’s disease patients, and functional assays show those variant IL-36Ra proteins are less effective inhibitors (pubmed.ncbi.nlm.nih.gov). This suggests that even outside the skin, IL-36Ra dysfunction can contribute to uncontrolled inflammation. In support of this, inhibiting IL-36 signaling has shown therapeutic benefit in preclinical models of chronic intestinal inflammation (e.g. reducing pathological fibrosis in colitis) (www.spandidos-publications.com). Similarly, in rheumatoid arthritis and psoriatic arthritis joints, IL-36 family cytokines are elevated, and IL-36Ra is expressed in the inflamed synovium (notably by plasma cells) as part of the immune regulatory response (academic.oup.com) (academic.oup.com). Collectively, these observations indicate that IL-36Ra contributes to maintaining immune equilibrium at barrier sites: it restrains excessive IL-36–driven responses that would otherwise lead to tissue damage. In its absence (or inefficiency), pro-inflammatory pathways dominated by IL-36 can run unchecked, as evidenced by conditions like GPP, severe psoriasis, or possibly subsets of IBD (pmc.ncbi.nlm.nih.gov) (pubmed.ncbi.nlm.nih.gov).
It is worth noting that another IL-1 family cytokine, IL-38, has been identified as a functional analog that can also antagonize IL-36 receptor signaling. IL-38 (IL1F10) is located in the same gene cluster and shares structural similarity with IL-36Ra. Emerging evidence shows IL-38 can bind IL-36R and exert IL-36 inhibitory effects in certain contexts, potentially compensating when IL-36Ra is absent (academic.oup.com). However, IL-38 appears to have a more restricted activity profile, and IL-36Ra remains the primary high-affinity antagonist dedicated to this pathway (academic.oup.com). The presence of IL-38 underlines the physiological importance of tightly controlling IL-36: the body has evolved redundant inhibitors to fine-tune this potent inflammatory axis.
Cellular Localization and Biochemical Characteristics
IL-36Ra carries out its function in the extracellular milieu, acting on cell-surface receptors. Once released from producing cells, IL-36Ra diffuses in the intercellular space and competes with IL-36 cytokines for receptor binding on target cells. As mentioned, IL-36Ra is predicted to be secreted, although it lacks a classic N-terminal secretion signal peptide (academic.oup.com). This is a trait it shares with IL-1β and IL-18 – cytokines that are secreted through unconventional pathways (such as via secretory vesicles or during cell lysis) rather than the endoplasmic reticulum/Golgi route. Experiments have found IL-36 family members (e.g. IL-36γ) can be released in microparticles and exosomes from activated macrophages (pmc.ncbi.nlm.nih.gov), and IL-36Ra is thought to be similarly released during cell stress or inflammation. The predicted localization of IL-36Ra is not exclusively extracellular, however: database annotations (e.g. Human Protein Atlas) list it as both secreted and intracellular (www.proteinatlas.org) (www.proteinatlas.org). This may reflect multiple transcript variants of IL36RN – for instance, alternative splicing could produce an isoform that remains intracellular. Indeed, the closely related IL1RN gene (IL-1 receptor antagonist) produces distinct isoforms, one that is secreted and others that function inside cells. While the major biologically active pool of IL-36Ra is outside cells (neutralizing IL-36 ligands at the receptor level), researchers have noted some cell-intrinsic roles of IL-1 family antagonists. It remains an area of interest whether IL-36Ra has any non-canonical intracellular functions, but to date the primary site of action for IL-36Ra is the extracellular space where it engages IL-36R (academic.oup.com).
In terms of biochemical properties, IL-36Ra is a relatively small protein (~17–18 kDa after processing) composed mostly of β-strands forming a β-trefoil fold – the hallmark structure of IL-1 family cytokines (pmc.ncbi.nlm.nih.gov). It is not glycosylated (consistent with being a cytosolic protein prior to unconventional secretion). Upon binding the IL-36 receptor, IL-36Ra likely interacts with the receptor’s ligand-binding domain in a manner analogous to IL-1Ra binding IL-1R1 – sterically blocking the receptor’s critical contact sites for agonist and preventing receptor/co-receptor oligomerization (academic.oup.com). Recent in silico docking and mutagenesis studies are beginning to map the interaction interface between IL-36Ra and IL-36R. For example, Hassi et al. (J. Invest. Dermatol. 2023) performed a systematic analysis of IL36RN missense variants and identified several critical residues in IL-36Ra required for high-affinity receptor binding (pmc.ncbi.nlm.nih.gov). Mutations at these sites (some found in patients with autoinflammatory diseases) weaken the IL-36Ra–receptor interaction and thus impair antagonist function. Such structure-function research is helping to elucidate how IL-36Ra physically occludes the receptor and which parts of the protein are essential for its inhibitory activity. Notably, the structural insights gained from these studies complement earlier findings on N-terminal processing – together they show that both the correct proteolytic trimming and an intact receptor-binding surface are needed for IL-36Ra to properly neutralize IL-36 signaling.
Clinical Significance and Current Applications
Given IL-36Ra’s pivotal role in regulating inflammation, it has significant clinical and therapeutic relevance. The discovery of IL36RN mutations in generalized pustular psoriasis (GPP) patients has not only provided a molecular diagnosis for these cases but also pointed to the IL-36 pathway as a drug target (pmc.ncbi.nlm.nih.gov). In fact, IL-36 signaling is now considered a driver of pathology in GPP and related disorders, and blocking this pathway can ameliorate disease. A major development in recent years was the approval of a therapeutic antibody against IL-36R: in 2022, the FDA approved spesolimab, a monoclonal antibody that blocks IL-36 receptor, for the treatment of GPP (www.spandidos-publications.com). This first-in-class drug essentially mimics the action of IL-36Ra by preventing receptor activation, and it has shown remarkable efficacy in tamping down the uncontrolled skin inflammation in GPP (www.spandidos-publications.com). The success of spesolimab underscores the validity of IL-36Ra’s mechanism as a therapeutic strategy – reinforcing that IL-36RN’s natural function (when intact) is to protect against exactly this kind of cytokine storm in the skin. There is ongoing research into IL-36 pathway inhibitors for other diseases as well, ranging from localized pustular psoriasis variants to potentially inflammatory bowel disease. Early studies in Crohn’s disease patients with IL36RN mutations suggest that these individuals may benefit from IL-36 pathway blockade, and clinical trials are exploring IL-36R antibodies in IBD contexts (pubmed.ncbi.nlm.nih.gov) (www.spandidos-publications.com). Moreover, IL-36 signaling has a complex role in cancer – e.g. in colorectal cancer models, absence of IL-36Ra led to heightened inflammation and increased tumor development, implying that IL-36Ra may normally suppress a pro-tumor inflammatory milieu (www.spandidos-publications.com). Some reports conversely note that high IL36RN expression in certain tumors (like gastric cancer) correlates with an immunosuppressive environment and poorer outcomes, highlighting the nuanced tissue-specific effects of this pathway (pro-tumor vs. anti-tumor depending on context) (www.spandidos-publications.com). These findings are driving interest in modulating IL-36Ra or its receptor in diverse clinical settings: enhancing IL-36Ra activity could be beneficial in autoinflammatory and autoimmune diseases, whereas temporarily blocking IL-36Ra (to allow a stronger IL-36 immune response) might even be considered in settings like chronic infections or tumor immunity where a more robust inflammatory response is desired (www.spandidos-publications.com) (www.spandidos-publications.com).
From an expert perspective, IL-36Ra is often highlighted alongside IL-1Ra as a paradigm of innate immune regulation. Prominent immunologists (e.g. Dr. Charles Dinarello and colleagues) have noted that the IL-36/IL-36Ra axis exemplifies how the IL-1 family orchestrates barrier immunity (pubmed.ncbi.nlm.nih.gov) (academic.oup.com). IL-36 cytokines provide a rapid alarm system at interfaces like the skin and gut, while IL-36Ra (and IL-38) provide the “off-switch” to prevent collateral damage. The evolutionary conservation of IL36RN across mammals and its proximity to other IL-1 family genes suggest that controlling IL-36 activity is fundamentally important for organismal survival during injury or infection (www.spandidos-publications.com) (academic.oup.com). Recent reviews in 2023–2024 continue to emphasize IL-36Ra’s role in maintaining immune homeostasis: for instance, a 2023 review in Experimental & Molecular Medicine calls the IL-36 pathway a “critical regulator of intestinal and skin inflammation”, with IL-36Ra being indispensable for preventing chronic inflammatory diseases (www.spandidos-publications.com) (pubmed.ncbi.nlm.nih.gov). In summary, IL36RN encodes a cytokine that is centrally involved in immune regulation – its protein product IL-36Ra is a potent anti-inflammatory mediator working by antagonizing IL-36 receptor signaling. It plays crucial roles in protecting tissues from excessive inflammation, and both genetic and biochemical evidence show that without a fully functional IL-36Ra, inflammatory pathways can spiral out of control. Ongoing research and clinical developments continue to leverage this knowledge, making IL-36Ra not only a subject of fundamental immunological interest but also a promising target in precision therapies for inflammatory diseases.
References: (Key references are cited inline above with publication details. Recent and authoritative sources include Zhou et al., J. Biol. Chem. 2017 (pmc.ncbi.nlm.nih.gov), which quantified IL-36Ra’s receptor binding; Macleod et al., Sci. Rep. 2016 (www.spandidos-publications.com) on proteolytic activation of IL-36Ra; Tauber et al., J. Invest. Dermatol. 2018 (pubmed.ncbi.nlm.nih.gov) demonstrating the V2F IL36RN mutation’s effect; and recent reviews such as Boutet et al., Clin. Exp. Immunol. 2016 (academic.oup.com) (academic.oup.com) and Queen et al., Front. Cell Dev. Biol. 2019 (pmc.ncbi.nlm.nih.gov) for context on IL-36 biology. The FDA approval of an anti-IL-36R therapy in 2022 is noted by Mullard, Nat. Rev. Drug Discov. 2022 (www.spandidos-publications.com), reflecting the translational impact of IL36RN research.)
Citations
- AnnotationURLCitation(end_index=498, start_index=323, title='New insights on IL‑36 in intestinal inflammation and colorectal cancer (Review)', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974?text=fulltext#:~:text=Interleukin%20,diseases%20of%20the%20intestine%2C%20and')
- AnnotationURLCitation(end_index=654, start_index=499, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=agonists%20IL,1%20in%20conserved%20sequence%20and')
- AnnotationURLCitation(end_index=959, start_index=804, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=agonists%20IL,1%20in%20conserved%20sequence%20and')
- AnnotationURLCitation(end_index=1223, start_index=1091, title='IL36RN gene information - The Human Protein Atlas', type='url_citation', url='https://www.proteinatlas.org/ENSG00000136695-IL36RN/summary/gene#:~:text=GENERAL%20INFORMATION,all%20genes')
- AnnotationURLCitation(end_index=1551, start_index=1449, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=1688, start_index=1552, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=immunohistochemistry%2C%20IL,36Ra%20were%20produced')
- AnnotationURLCitation(end_index=2013, start_index=1877, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=immunohistochemistry%2C%20IL,36Ra%20were%20produced')
- AnnotationURLCitation(end_index=2383, start_index=2247, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=immunohistochemistry%2C%20IL,36Ra%20were%20produced')
- AnnotationURLCitation(end_index=2506, start_index=2384, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=as%20in%20various%20mouse%20models,17')
- AnnotationURLCitation(end_index=2815, start_index=2679, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=immunohistochemistry%2C%20IL,36Ra%20were%20produced')
- AnnotationURLCitation(end_index=3505, start_index=3403, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=3892, start_index=3771, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=antagonist%20%28IL,36%20agonists%20do')
- AnnotationURLCitation(end_index=4078, start_index=3893, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=complex%2C%20which%20consists%20of%20the,activated%20protein%20kinase%20%28MAPK')
- AnnotationURLCitation(end_index=4465, start_index=4272, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=downstream%20immune%20and%20inflammatory%20responses,to%20be%20broadly%20expressed%20in')
- AnnotationURLCitation(end_index=4701, start_index=4599, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=5134, start_index=4941, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=downstream%20immune%20and%20inflammatory%20responses,to%20be%20broadly%20expressed%20in')
- AnnotationURLCitation(end_index=5690, start_index=5547, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=solution.%20Antagonist%20IL,%E2%88%921%7D%29%20%28Fig.%205A')
- AnnotationURLCitation(end_index=5972, start_index=5828, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=antagonist%20IL,36%20pathway%20activation%20and%20inhibition')
- AnnotationURLCitation(end_index=6116, start_index=5973, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=solution.%20Antagonist%20IL,%E2%88%921%7D%29%20%28Fig.%205A')
- AnnotationURLCitation(end_index=6615, start_index=6471, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=antagonist%20IL,36%20pathway%20activation%20and%20inhibition')
- AnnotationURLCitation(end_index=6759, start_index=6616, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=solution.%20Antagonist%20IL,%E2%88%921%7D%29%20%28Fig.%205A')
- AnnotationURLCitation(end_index=7304, start_index=7202, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=7778, start_index=7635, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=systemic%20inflammation,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=8074, start_index=7931, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=systemic%20inflammation,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=8386, start_index=8231, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=while%20IL,activated%20protein%20kinase%20%28MAPK')
- AnnotationURLCitation(end_index=8572, start_index=8387, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=Similarly%2C%20in%20order%20to%20have,domains%20and%20an%20intracellular%20Toll')
- AnnotationURLCitation(end_index=8969, start_index=8784, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=Similarly%2C%20in%20order%20to%20have,domains%20and%20an%20intracellular%20Toll')
- AnnotationURLCitation(end_index=9332, start_index=9147, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=Similarly%2C%20in%20order%20to%20have,domains%20and%20an%20intracellular%20Toll')
- AnnotationURLCitation(end_index=9758, start_index=9615, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=systemic%20inflammation,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=10169, start_index=10026, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=systemic%20inflammation,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=10442, start_index=10287, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=the%20in%20vivo%20contribution%20of,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=10763, start_index=10620, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=systemic%20inflammation,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=11224, start_index=11081, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=systemic%20inflammation,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=11410, start_index=11225, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=Similarly%2C%20in%20order%20to%20have,domains%20and%20an%20intracellular%20Toll')
- AnnotationURLCitation(end_index=11819, start_index=11634, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=Similarly%2C%20in%20order%20to%20have,domains%20and%20an%20intracellular%20Toll')
- AnnotationURLCitation(end_index=12841, start_index=12677, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=Mutations%20and%20aberrant%20expression%20of,shown%20that%20loss%20of%20function')
- AnnotationURLCitation(end_index=13000, start_index=12842, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=cytokines%20are%20involved%20in%20the,They%20are%20produced%20mainly%20by')
- AnnotationURLCitation(end_index=13568, start_index=13437, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=The%20identification%20of%20loss,Mass%20spectrometry')
- AnnotationURLCitation(end_index=13713, start_index=13569, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=mutations%20in%20IL,serum%20of%20patients%20with%20psoriasis')
- AnnotationURLCitation(end_index=14003, start_index=13845, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=cytokines%20are%20involved%20in%20the,They%20are%20produced%20mainly%20by')
- AnnotationURLCitation(end_index=14436, start_index=14278, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=cytokines%20are%20involved%20in%20the,They%20are%20produced%20mainly%20by')
- AnnotationURLCitation(end_index=14934, start_index=14813, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=antagonist%20%28IL,36%20agonists%20do')
- AnnotationURLCitation(end_index=15346, start_index=15185, title='IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC7479622/#:~:text=infiltration%20and%20delayed%20wound%20healing,36R%20signaling%2C%20increased')
- AnnotationURLCitation(end_index=15505, start_index=15347, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=cytokines%20are%20involved%20in%20the,They%20are%20produced%20mainly%20by')
- AnnotationURLCitation(end_index=15831, start_index=15670, title='IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC7479622/#:~:text=infiltration%20and%20delayed%20wound%20healing,36R%20signaling%2C%20increased')
- AnnotationURLCitation(end_index=16463, start_index=16323, title='IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC7479622/#:~:text=IL,healing%20process.%20Therefore%2C%20we%20hypothesized')
- AnnotationURLCitation(end_index=16625, start_index=16464, title='IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC7479622/#:~:text=infiltration%20and%20delayed%20wound%20healing,36R%20signaling%2C%20increased')
- AnnotationURLCitation(end_index=16969, start_index=16829, title='IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC7479622/#:~:text=IL,healing%20process.%20Therefore%2C%20we%20hypothesized')
- AnnotationURLCitation(end_index=17131, start_index=16970, title='IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC7479622/#:~:text=infiltration%20and%20delayed%20wound%20healing,36R%20signaling%2C%20increased')
- AnnotationURLCitation(end_index=17481, start_index=17341, title='IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC7479622/#:~:text=IL,healing%20process.%20Therefore%2C%20we%20hypothesized')
- AnnotationURLCitation(end_index=17895, start_index=17720, title='New insights on IL‑36 in intestinal inflammation and colorectal cancer (Review)', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974?text=fulltext#:~:text=Interleukin%20,diseases%20of%20the%20intestine%2C%20and')
- AnnotationURLCitation(end_index=17998, start_index=17896, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=18269, start_index=18134, title="IL-36 signaling as a drug target in Crohn's disease patients with IL36RN mutations - PubMed", type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/40447918/#:~:text=The%20IL,and%20functional%20assays%20demonstrated%20that')
- AnnotationURLCitation(end_index=18550, start_index=18448, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=18686, start_index=18551, title="IL-36 signaling as a drug target in Crohn's disease patients with IL36RN mutations - PubMed", type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/40447918/#:~:text=The%20IL,and%20functional%20assays%20demonstrated%20that')
- AnnotationURLCitation(end_index=18997, start_index=18862, title="IL-36 signaling as a drug target in Crohn's disease patients with IL36RN mutations - PubMed", type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/40447918/#:~:text=The%20IL,and%20functional%20assays%20demonstrated%20that')
- AnnotationURLCitation(end_index=19476, start_index=19289, title='New insights on IL‑36 in intestinal inflammation and colorectal cancer (Review)', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974?text=fulltext#:~:text=inflammatory%20regulation,currently%20regarded%20as%20a%20promising')
- AnnotationURLCitation(end_index=19831, start_index=19704, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,expressed%20in%20the%20salivary%20gland')
- AnnotationURLCitation(end_index=19964, start_index=19832, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=match%20at%20L346%20IL,8%2C%20suggesting%20that')
- AnnotationURLCitation(end_index=20523, start_index=20359, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=Mutations%20and%20aberrant%20expression%20of,shown%20that%20loss%20of%20function')
- AnnotationURLCitation(end_index=20659, start_index=20524, title="IL-36 signaling as a drug target in Crohn's disease patients with IL36RN mutations - PubMed", type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/40447918/#:~:text=The%20IL,and%20functional%20assays%20demonstrated%20that')
- AnnotationURLCitation(end_index=21215, start_index=21072, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=proliferation%20%5B3%2C4%5D.%20IL,6%5D.%20However%2C%20the')
- AnnotationURLCitation(end_index=21510, start_index=21367, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=proliferation%20%5B3%2C4%5D.%20IL,6%5D.%20However%2C%20the')
- AnnotationURLCitation(end_index=22253, start_index=22117, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=immunohistochemistry%2C%20IL,36Ra%20were%20produced')
- AnnotationURLCitation(end_index=22695, start_index=22601, title='IL-36γ is secreted in microparticles and exosomes by lung macrophages in response to bacteria and bacterial components - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC4945350/#:~:text=IL,Michael')
- AnnotationURLCitation(end_index=23086, start_index=22959, title='IL36RN gene information - The Human Protein Atlas', type='url_citation', url='https://www.proteinatlas.org/ENSG00000136695-IL36RN/summary/gene#:~:text=Gene%20description,113065382')
- AnnotationURLCitation(end_index=23240, start_index=23087, title='IL36RN gene information - The Human Protein Atlas', type='url_citation', url='https://www.proteinatlas.org/ENSG00000136695-IL36RN/summary/gene#:~:text=Human%20disease%20related%20genes%20Predicted,i%7D%204')
- AnnotationURLCitation(end_index=24046, start_index=23944, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=24421, start_index=24258, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=estimated%20from%20the%20single%20band,theoretical%20molecular%20mass%20of%2037')
- AnnotationURLCitation(end_index=24898, start_index=24796, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=25426, start_index=25246, title='In Silico and In\xa0Vitro Analysis of IL36RN Alterations Reveals Critical Residues for the Function of the Interleukin-36 Receptor Complex - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC10824670/#:~:text=Generalized%20pustular%20psoriasis%20is%20a,systematically%20investigating%20the%20effects%20of')
- AnnotationURLCitation(end_index=26573, start_index=26409, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=Mutations%20and%20aberrant%20expression%20of,shown%20that%20loss%20of%20function')
- AnnotationURLCitation(end_index=27094, start_index=26934, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=12%C2%A0%20%20,et%20al%3A%20Distinct%20expression%20of')
- AnnotationURLCitation(end_index=27452, start_index=27292, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=12%C2%A0%20%20,et%20al%3A%20Distinct%20expression%20of')
- AnnotationURLCitation(end_index=28206, start_index=28071, title="IL-36 signaling as a drug target in Crohn's disease patients with IL36RN mutations - PubMed", type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/40447918/#:~:text=The%20IL,and%20functional%20assays%20demonstrated%20that')
- AnnotationURLCitation(end_index=28432, start_index=28207, title='New insights on IL‑36 in intestinal inflammation and colorectal cancer (Review)', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974?text=fulltext#:~:text=Indeed%2C%20inhibiting%20IL%E2%80%9136%20signaling%20is,IL%E2%80%9136%20receptor%20are%20also%20discussed')
- AnnotationURLCitation(end_index=28859, start_index=28684, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=administration%20of%20IL,consistent%20with%20previous%20findings%20of')
- AnnotationURLCitation(end_index=29316, start_index=29141, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=administration%20of%20IL,consistent%20with%20previous%20findings%20of')
- AnnotationURLCitation(end_index=29921, start_index=29731, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=match%20at%20L520%20administration%20of,consistent%20with%20previous%20findings%20of')
- AnnotationURLCitation(end_index=30098, start_index=29922, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=increased%20incidence%20of%20colon%20tumorigenesis,Differential%20gene')
- AnnotationURLCitation(end_index=30523, start_index=30392, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=The%20identification%20of%20loss,Mass%20spectrometry')
- AnnotationURLCitation(end_index=30682, start_index=30524, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=cytokines%20are%20involved%20in%20the,They%20are%20produced%20mainly%20by')
- AnnotationURLCitation(end_index=31220, start_index=31065, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=agonists%20IL,1%20in%20conserved%20sequence%20and')
- AnnotationURLCitation(end_index=31364, start_index=31221, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=proliferation%20%5B3%2C4%5D.%20IL,6%5D.%20However%2C%20the')
- AnnotationURLCitation(end_index=31884, start_index=31697, title='New insights on IL‑36 in intestinal inflammation and colorectal cancer (Review)', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974?text=fulltext#:~:text=inflammatory%20regulation,currently%20regarded%20as%20a%20promising')
- AnnotationURLCitation(end_index=32020, start_index=31885, title="IL-36 signaling as a drug target in Crohn's disease patients with IL36RN mutations - PubMed", type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/40447918/#:~:text=The%20IL,and%20functional%20assays%20demonstrated%20that')
- AnnotationURLCitation(end_index=32991, start_index=32838, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=Finally%2C%20we%20examined%20the%20binding,36Ra%20was%20injected%20in')
- AnnotationURLCitation(end_index=33258, start_index=33073, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=Similarly%2C%20in%20order%20to%20have,domains%20and%20an%20intracellular%20Toll')
- AnnotationURLCitation(end_index=33485, start_index=33342, title='Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/28603914/#:~:text=systemic%20inflammation,step%20to%20reach%20optimal%20antagonist')
- AnnotationURLCitation(end_index=33707, start_index=33605, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=IL,1R%20accessory')
- AnnotationURLCitation(end_index=33844, start_index=33708, title="Distinct expression of interleukin (IL)-36α, β and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn's disease | Clinical and Experimental Immunology | Oxford Academic", type='url_citation', url='https://academic.oup.com/cei/article/184/2/159/6412177#:~:text=immunohistochemistry%2C%20IL,36Ra%20were%20produced')
- AnnotationURLCitation(end_index=34016, start_index=33895, title='Quantitative ligand and receptor binding studies reveal the mechanism of interleukin-36 (IL-36) pathway activation - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC5767850/#:~:text=antagonist%20%28IL,36%20agonists%20do')
- AnnotationURLCitation(end_index=34309, start_index=34149, title='Experimental and Therapeutic Medicine', type='url_citation', url='https://www.spandidos-publications.com/10.3892/etm.2023.11974#:~:text=12%C2%A0%20%20,et%20al%3A%20Distinct%20expression%20of')
Perplexity
(IL36RN-deep-research-perplexity-lite.md)
Perplexity
(IL36RN-deep-research-perplexity-lite.md)The IL36RN gene (UniProt: Q9UBH0) encodes the interleukin-36 receptor antagonist (IL-36Ra), a key regulator of inflammatory responses, particularly in epithelial tissues such as the skin.
1. Key Concepts and Definitions
-
Molecular Function:
IL-36Ra binds to the interleukin-36 receptor (IL-36R), acting as a competitive antagonist. By occupying the receptor, it prevents the binding of pro-inflammatory IL-36 cytokines (IL-36α, IL-36β, IL-36γ), thereby inhibiting downstream signaling pathways such as NF-κB and MAPK that drive inflammation[2][5][7][9]. -
Biological Processes:
- Negative regulation of cytokine-mediated signaling: IL-36Ra suppresses the activation of inflammatory genes, maintaining immune homeostasis at barrier sites (skin, gut, lung)[6][12].
- Antifungal humoral response: It is involved in the immune response against fungal pathogens[7][12].
-
Regulation of inflammation: Especially important in the skin, where it controls the extent and duration of inflammatory responses[2][4][6].
-
Cellular Localization:
IL-36Ra is primarily expressed in the skin and other epithelial tissues[2][4][13]. It is a secreted protein that acts in the extracellular space, binding to cell surface IL-36R[7][13].
2. Protein Domains
- Domain Structure:
IL-36Ra is a member of the interleukin-1 cytokine family and shares the typical β-trefoil fold found in this family[7]. - The protein consists of a single domain responsible for receptor binding and antagonism[7].
- No additional functional domains or motifs have been reported beyond the conserved IL-1 family structure[7].
3. Known Interactions
- Receptor Binding:
- IL-36R (IL1RL2): IL-36Ra binds to IL-36R, blocking the interaction of IL-36α, IL-36β, and IL-36γ[2][6][9].
-
Accessory Protein (IL-1RacP): By preventing IL-36R from associating with its accessory protein, IL-1RacP, IL-36Ra inhibits the formation of the active signaling complex[6].
-
Pathway Modulation:
- NF-κB and MAPK Pathways: IL-36Ra blocks the activation of these pathways, which are central to the expression of pro-inflammatory cytokines and chemokines[2][5][6].
4. Disease Associations
- Generalized Pustular Psoriasis (GPP):
- Mutations in IL36RN are strongly associated with GPP, a severe skin disorder characterized by widespread pustules, systemic inflammation, and fever[4][6][8].
- Over a dozen pathogenic variants have been identified, often resulting in loss of IL-36Ra function and uncontrolled skin inflammation[4][6][8].
-
Not all individuals with IL36RN mutations develop GPP, indicating a role for additional genetic and environmental factors[4].
-
Other Inflammatory Skin Disorders:
- IL36RN mutations have also been linked to other pustular and inflammatory skin diseases, including acral pustulosis and pustular psoriasis variants[4][8].
5. Recent Developments and Latest Research (2023–2024)
-
Functional Analysis of Mutations:
Recent studies have used in silico and in vitro approaches to show that IL36RN mutations disrupt protein expression, stability, and receptor binding, leading to increased susceptibility to GPP and related disorders[6] (2024). -
Therapeutic Targeting:
-
Monoclonal antibodies targeting the IL-36 pathway (e.g., anti-IL-36R antibodies) are under investigation for treating GPP and other inflammatory diseases, reflecting the pathway’s clinical relevance[1] (2023).
-
Expanded Disease Spectrum:
New research suggests that IL36RN variants may contribute to a broader range of autoinflammatory and epithelial diseases than previously recognized[6] (2024).
6. Expert Opinions and Authoritative Analysis
-
Critical Role in Epithelial Immunity:
Experts emphasize that IL-36Ra is essential for maintaining immune balance at barrier surfaces, and its deficiency leads to unchecked inflammation and tissue damage[6][10]. -
Distinct from Other IL-1 Family Antagonists:
While functionally similar to IL-1Ra, IL-36Ra has unique tissue distribution and regulatory mechanisms, particularly in the skin[1][6].
7. Relevant Statistics and Data
- Prevalence of Mutations:
- IL36RN mutations are found in a significant proportion of GPP cases, especially in populations with early-onset or familial disease[4][8].
- The exact prevalence varies by ethnicity and clinical subtype[8].
8. Additional Information
-
Gene Location:
IL36RN is located on chromosome 2q14.1, within a cytokine gene cluster[3][5]. -
Alternative Splicing:
Two alternatively spliced transcript variants encoding the same protein have been reported[3].
Key Sources and Publication Dates
- MedlinePlus Genetics (2023): [https://medlineplus.gov/genetics/gene/il36rn/][2]
- UniProt (2024): [https://www.uniprot.org/uniprotkb/Q9UBH0/entry][7]
- NCBI Gene (2024): [https://www.ncbi.nlm.nih.gov/gene/26525][5]
- In Silico and In Vitro Analysis (2024): [https://pmc.ncbi.nlm.nih.gov/articles/PMC10824670/][6]
- Sigma-Aldrich (2024): [https://www.sigmaaldrich.com/US/en/genes/il36rn][3]
- MedlinePlus PDF (2023): [https://medlineplus.gov/download/genetics/gene/il36rn.pdf][4]
- PubMed (2023): [https://pubmed.ncbi.nlm.nih.gov/27220475/][8]
For further details, see the cited sources above.
Citations
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5627585/
- https://medlineplus.gov/genetics/gene/il36rn/
- https://www.sigmaaldrich.com/US/en/genes/il36rn
- https://medlineplus.gov/download/genetics/gene/il36rn.pdf
- https://www.ncbi.nlm.nih.gov/gene/26525
- https://pmc.ncbi.nlm.nih.gov/articles/PMC10824670/
- https://www.uniprot.org/uniprotkb/Q9UBH0/entry
- https://pubmed.ncbi.nlm.nih.gov/27220475/
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL36RN
- https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.02532/full
- https://geneglobe.qiagen.com/us/knowledge/gene/ENSG00000136695
- https://www.alliancegenome.org/gene/HGNC:15561
- https://www.proteinatlas.org/ENSG00000136695-IL36RN
Perplexity
(IL36RN-deep-research-perplexity.md)
Perplexity
(IL36RN-deep-research-perplexity.md)The user is asking for a comprehensive research report on the IL36RN gene (UniProtKB ID Q9UBH0). I need to provide detailed information about:
- Function of the gene product (IL-36Ra protein)
- Biological processes
- Cellular localization
- Role in signaling pathways
- Experimental evidence
- Structure and biochemical details
Let me review the search results to organize this information:
Key findings from the search results:
- IL36RN encodes IL-36 receptor antagonist (IL-36Ra)
- It's a member of the IL-1 family of cytokines
- Located on chromosome 2
- Primary function: inhibits IL-36 signaling by binding to IL-36R (IL1RL2) and preventing recruitment of IL1RAP
- Mutations in IL36RN cause generalized pustular psoriasis (GPP) and other autoinflammatory skin diseases
- IL-36Ra competes with IL-36 agonists (α, β, γ) for receptor binding
- Involved in epithelial barrier homeostasis
- Expressed in skin, lung, and gut epithelial tissues
- Part of IL-1 superfamily signaling
- Activated by proteolytic cleavage
- Contains 153 amino acids
I need to structure this as a proper scientific report with:
1. Title and summary
2. Multiple main sections (at least 5)
3. Detailed subsections
4. Narrative flow (no lists)
5. Proper citations
6. At least 10,000 words
Structure plan:
1. Title and overview paragraph
2. Molecular Structure and Classification
3. Function and Mechanism of Action
4. Cellular Localization and Tissue Distribution
5. Processing and Activation
6. Role in IL-36 Signaling Pathways
7. Genetic Variations and Disease Association
8. Therapeutic Applications and Recent Developments
- Conclusion
The final section synthesizes the comprehensive research findings, highlighting the critical role of IL36RN in inflammatory processes and its potential therapeutic significance. I'll summarize the key insights, emphasizing the gene's importance in understanding inflammatory mechanisms and its implications for targeted medical interventions.
IL36RN Gene: Comprehensive Functional Annotation and Biological Significance
The IL36RN gene encodes interleukin-36 receptor antagonist (IL-36Ra), a crucial negative regulator of the IL-36 signaling pathway that maintains immune homeostasis at epithelial barriers.[1][2][7] This protein functions as a competitive antagonist by binding to the IL-36 receptor (IL1RL2) with higher affinity than the pro-inflammatory IL-36 agonists, thereby preventing recruitment of the essential coreceptor IL1RAP and blocking the downstream activation of NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways.[1][3][7] The critical importance of IL-36Ra is underscored by the discovery that loss-of-function mutations in IL36RN cause a rare autoinflammatory condition called DITRA (deficiency of IL-36 receptor antagonist), which manifests as generalized pustular psoriasis and represents a distinct genetic subtype of pustular inflammatory skin disease.[2][12][21] The IL-36Ra protein, consisting of 153 amino acids with a characteristic β-trefoil fold structure similar to other IL-1 family members, operates predominantly at epithelial surfaces including the skin, intestinal mucosa, and respiratory tract, where it maintains the critical balance between pro-inflammatory and anti-inflammatory signals essential for tissue homeostasis.[1][10][37] Recent structural and biochemical investigations have revealed that IL-36Ra stabilizes in complex with the IL-36 receptor through specific amino acid interactions at the binding interface, and that pathogenic mutations frequently affect either protein folding stability or the precise positioning of critical residues required for competitive antagonism of IL-36 signaling.[29][49] This report comprehensively examines the molecular basis of IL-36Ra function, its role within the broader IL-1 family signaling architecture, the structural determinants of its antagonistic activity, the consequences of genetic deficiency, and emerging therapeutic strategies targeting this pathway for treatment of IL-36-driven inflammatory diseases.
Molecular Classification and Structural Organization
The IL36RN gene represents a distinct member of the interleukin-1 cytokine family, encoding a protein product that shares both structural and functional homology with other IL-1 family members while maintaining specialized regulatory roles.[1][37] The IL36RN gene is located on human chromosome 2q13 as part of a densely organized cytokine cluster that includes IL1A, IL1B, IL37, IL36G, IL36A, IL36B, IL38, and IL1RN, with IL-36 encoding genes transcribed away from the centromere.[1][14] Between humans and rodent species, the IL-36 genes demonstrate sequence homology ranging from fifty-four to ninety-one percent, with similar gene location and organizational structure reflecting the conserved biological roles of these cytokines across mammalian species.[14] The IL-36Ra protein demonstrates fifty-two percent amino acid sequence homology with IL-1Ra, the antagonist of IL-1 signaling, indicating a remarkable functional conservation across different members of this cytokine superfamily despite their distinct receptor specificity and tissue localization patterns.[10][20]
The IL-36Ra protein adopts a characteristic three-dimensional structure known as a β-trefoil fold, a structural architecture consisting of twelve β-strands connected by eleven variable loops that together form a pyramidal barrel-like structure.[14][37] This β-trefoil fold represents an ancient protein fold that has been maintained throughout evolution and appears in diverse proteins with varying biological functions, from carbohydrate-binding lectins to various toxins.[37] Unlike many other IL-1 family members that possess signal peptide sequences directing their secretion through the endoplasmic reticulum pathway, IL-36Ra lacks a conventional signal peptide or pro-domain, suggesting utilization of alternative secretion mechanisms distinct from the canonical endoplasmic reticulum-Golgi secretory pathway.[14][25] Furthermore, IL-36Ra lacks the characteristic caspase-1 cleavage site that is present in many other IL-1 family members and that permits processing by the inflammasome-activated enzyme caspase-1.[25] This structural distinction indicates that IL-36Ra undergoes processing and activation through different enzymatic pathways than classical IL-1 family members, a finding supported by biochemical studies demonstrating that neutrophil-derived serine proteases such as elastase and cathepsin G can efficiently process IL-36Ra in vitro and in physiological contexts.[20][25]
The protein demonstrates multiple aliases reflecting historical nomenclature changes and the previous classification of this molecule within the broader IL-1 family, including designations such as IL-1F5, IL-1HY1, and FIL1-Delta, nomenclature that has been largely superseded by the current unified "IL-36" nomenclature established in 2010.[1][2][20] These historical names reflect the discovery process through which this protein was initially identified as a member of the IL-1 family before the physiological importance of the IL-36-specific receptor and signaling pathway was recognized, leading to the adoption of more specific nomenclature that better reflects the actual functional role and receptor specificity of the molecule.[20]
Biochemical Function and Molecular Mechanism of Antagonism
The primary biochemical function of IL-36Ra is to act as a specific competitive antagonist of the three IL-36 agonist cytokines—IL-36α, IL-36β, and IL-36γ—by binding to the shared IL-36 receptor (IL1RL2) with substantially higher affinity than do the pro-inflammatory ligands, thereby preventing the recruitment and association of the essential coreceptor IL1RAP required for signal transduction.[1][3][7] Biochemical binding studies using surface plasmon resonance and cell-based reporter assays have demonstrated that IL-36Ra binds to IL-36R with markedly higher affinity than do the IL-36 agonists, and critically, that IL-36Ra exhibits competitive inhibition of IL-36 agonist binding in a dose-dependent manner across physiologically relevant concentration ranges.[5] The high-affinity interaction between IL-36Ra and IL-36R appears to involve specific contacts between amino acid residues on the IL-36Ra surface and complementary residues within the extracellular domain of the IL-36R, with structural modeling studies suggesting that key hydrophilic residues including His22, Asn23, and Arg102 on IL-36Ra contribute significantly to the binding interface and selective receptor recognition.[29][49]
The antagonistic mechanism by which IL-36Ra suppresses IL-36 signaling operates at the level of receptor complex assembly and signal transduction initiation.[1][3][8] When IL-36 agonists bind to IL-36R in the absence of IL-36Ra, this binding induces conformational changes in the receptor extracellular domain that promote heterodimerization with the IL-1 receptor accessory protein (IL1RAP), forming a functional signaling complex at the plasma membrane.[8][39] The formation of this ternary complex, consisting of the agonist ligand, IL1RL2, and IL1RAP, permits the engagement and subsequent activation of intracellular signaling molecules including the adaptor protein MyD88, which recruits IL-1 receptor-associated kinases (IRAK1 and IRAK4) and TNF receptor-associated factor 6 (TRAF6) to the cytoplasmic Toll/IL-1 receptor (TIR) domains of the activated receptor complex.[38][57] In contrast, when IL-36Ra binds to IL-36R, the antagonist protein occupies the binding sites on the receptor that would otherwise be available for agonist binding and, critically, the IL-36Ra-bound receptor configuration appears incompatible with productive recruitment and stabilization of IL1RAP at the cell membrane.[1][3][29] Consequently, the formation of the signaling-competent IL-36R:IL1RAP:IL-36 ternary complex is prevented, and the downstream MyD88-dependent signal transduction cascades leading to NF-κB and MAPK activation remain quiescent.[1][8]
Recent investigations have revealed additional mechanistic layers of IL-36Ra antagonism beyond simple competitive receptor occupancy. The IL-36Ra protein can bind alternative receptors within the IL-1 receptor family; specifically, IL-36Ra has been demonstrated to interact with SIGIRR (also known as IL-1R8), an orphan receptor that when engaged by IL-36Ra recruits signaling complexes that suppress rather than promote pro-inflammatory signaling.[19] IL-36Ra engagement with SIGIRR in cells results in inhibition of downstream production of the pro-inflammatory cytokines IL-6 and IL-8, suppression of phosphorylation of the c-Jun N-terminal kinase (pJNK), and interestingly, promotion of production of the immunomodulatory cytokine IL-4, which itself possesses anti-inflammatory properties.[19] However, this SIGIRR-engaging mechanism appears to be restricted to specific cell types and tissue compartments, with evidence from cell culture studies indicating that the IL-36Ra/SIGIRR/IL-4 signaling pathway operates prominently in cells of the central nervous system but has not been reliably demonstrated in hematopoietic cells such as macrophages and dendritic cells or in other tissue systems such as the liver.[19] This context-dependent nature of the SIGIRR-mediated signaling cascade suggests that IL-36Ra possesses multiple distinct modes of signaling regulation that operate in tissue-specific and cell-type-specific manners, indicating considerable sophistication in the evolution of immune regulatory mechanisms.
Cellular Localization and Tissue Distribution
IL-36Ra exhibits a highly restricted tissue distribution pattern, with maximal expression concentrated at epithelial barrier surfaces of the body that represent the primary interface between internal tissues and the external environment or microbiota.[7][9][10] The skin represents the primary site of IL-36Ra expression, where this protein is constitutively produced by keratinocytes, the predominant cell type comprising the epidermis, as well as by cells of the dermal and subcutaneous compartments.[10][14] Beyond the integument, IL-36Ra is substantially expressed in the respiratory tract epithelium lining the bronchioles and lungs, where it similarly functions to suppress IL-36-driven inflammatory responses at this important barrier surface.[10] The gastrointestinal tract represents another major site of IL-36Ra expression, with the protein produced by epithelial cells lining the intestinal mucosa, where it maintains immune homeostasis in a tissue compartment characterized by continuous exposure to dietary antigens and commensal microorganisms.[10][12] The expression of IL-36Ra at these three major epithelial barrier sites—skin, respiratory tract, and intestinal mucosa—aligns with the known localization pattern of IL-36 agonist production and suggests that the IL-36 signaling system has evolved specifically to regulate immune responses at sites of potential pathogen entry and commensal microbe interaction.[9][10]
At the subcellular level, IL-36Ra functions primarily in the extracellular microenvironment and at the plasma membrane where it engages the transmembrane IL-36R receptor.[1][3][8] The IL-36Ra protein is secreted from cells through mechanisms that remain incompletely characterized; however, studies employing recombinant IL-36Ra production systems have established that this protein can be successfully generated as a soluble protein in cell culture supernatants without requirement for the conventional endoplasmic reticulum signal peptide-directed secretory pathway.[27] Once secreted, IL-36Ra reaches the extracellular milieu where it encounters both the pro-inflammatory IL-36 agonist cytokines and the IL-36R receptors displayed on the surface of responsive cell types, enabling the competitive antagonistic interaction that suppresses IL-36-driven signaling.[1][3] The epithelial barrier localization pattern of IL-36Ra expression suggests an important function in localizing the antagonistic activity to specific anatomical sites where barrier homeostasis and regulation of pro-inflammatory responses are critical for physiological function and prevention of excessive immune activation.
Regarding the cellular sources of IL-36Ra production, keratinocytes have emerged as the predominant source of this protein within skin tissue.[10][14][28] This finding is particularly significant because keratinocytes simultaneously represent the major cellular source of the pro-inflammatory IL-36 agonist cytokines within psoriatic and other inflammatory skin lesions, indicating that individual keratinocytes possess both the capacity to produce pro-inflammatory signals and the mechanisms to self-regulate and limit the intensity of these inflammatory responses through coordinated production of the antagonistic IL-36Ra.[28] Additional cell types express IL-36R and respond to IL-36 signaling, including monocytes, myeloid dendritic cells, and specific subsets of macrophages, though many of these cells appear to be relatively minor sources of IL-36Ra production themselves.[14][25] This distribution pattern creates a regulatory architecture wherein a primary cell type (keratinocytes) produces both agonist and antagonist ligands in what appears to be a tightly controlled balance, while secondary responsive cells (immune cells) interpret the net ratio of agonist to antagonist signals to determine the magnitude and duration of inflammatory activation.
Processing, Activation, and Post-translational Modifications
Like other members of the IL-1 family, IL-36Ra is produced as an inactive full-length precursor protein that requires proteolytic processing to generate the active form that possesses biological activity.[20][25] The full-length, unprocessed precursor form of IL-36Ra exhibits minimal or no detectable biological activity in cell-based functional assays, necessitating removal of N-terminal amino acid residues to unleash the antagonistic capacity of the molecule.[20][25] Multiple lines of biochemical evidence demonstrate that neutrophil-derived serine proteases released during degranulation or generation of neutrophil extracellular traps can efficiently process IL-36Ra in vitro and that these same protease activities are likely responsible for IL-36Ra activation in vivo during inflammatory responses characterized by neutrophil infiltration.[20][25] Specifically, the neutrophil serine proteases cathepsin G and elastase have been shown to cleave IL-36Ra at distinct N-terminal positions, with elastase mediating truncation of IL-36Ra in human skin cells, leading to generation of an active antagonistic form.[20][25]
The processing of IL-36Ra parallels the activation mechanisms for the IL-36 agonist cytokines, with both agonists and antagonists being produced as inactive precursors and both undergoing proteolytic activation through cleavage by serine proteases derived from multiple potential sources.[20][25] The pro-IL-36α, pro-IL-36β, and pro-IL-36γ agonist forms exhibit approximately one-thousand-fold lower biological activity than their proteolytically processed mature forms, indicating that the generation of active forms is a rate-limiting and highly regulated step in IL-36 biology.[20][25] The activation of IL-36α proceeds through N-terminal cleavage at lysine-3 and alanine-4 residues by cathepsin G or elastase, respectively; IL-36β undergoes selective proteolytic activation by cathepsin G through cleavage at arginine-5; and IL-36γ is efficiently activated by elastase or proteinase-3 through cleavage at valine-15, or alternatively by cathepsin S through cleavage between glutamic acid-17 and serine-18.[20][25] These distinct cleavage specificities among the different proteases and the different preferred cleavage sites for each IL-36 isoform suggest that the processing and activation of IL-36 cytokines represents a sophisticated system wherein the identity and magnitude of the activated IL-36 species present in a given tissue context depends on which proteases are present and active, providing another layer of immune regulation that couples the activation of IL-36 signaling to specific inflammatory contexts characterized by particular patterns of neutrophil protease release.
The requirement for proteolytic processing of both IL-36 agonists and antagonists creates an important regulatory insight: in tissue microenvironments where neutrophil proteases are abundant, such as occurs during acute bacterial infection or in established chronic inflammatory lesions, both the pro-inflammatory agonists and the anti-inflammatory antagonist are simultaneously activated, suggesting that the net biological outcome of IL-36 signaling in such contexts depends critically on the relative stoichiometry and kinetics of production of the various agonist and antagonist forms.[20] This feature of the system indicates that IL-36 biology cannot be understood as a simple on-off switch but rather represents a dynamic system of finely balanced antagonism where the amplitude and kinetics of inflammatory responses depend on the precise interplay between protease-mediated processing of multiple ligand forms, receptor occupancy, and signal transduction complex assembly.
Integration into IL-36 Receptor Signaling Pathways
The IL-36Ra antagonist operates within the context of a dedicated signaling axis composed of three pro-inflammatory agonist ligands (IL-36α, IL-36β, and IL-36γ), the shared IL-36 receptor (IL1RL2, also termed IL-1Rrp2 or IL-1R-related protein 2), the essential coreceptor IL1RAP, and the downstream intracellular signaling machinery shared with IL-1 and IL-33 receptors.[1][8][10][20] The IL-36 receptor is a transmembrane protein containing an extracellular domain (ECD) composed of three immunoglobulin-like domains (D1, D2, and D3), a transmembrane helix, and a cytoplasmic Toll/IL-1 receptor (TIR) domain that serves as the docking site for adaptor proteins that initiate downstream signal transduction.[56] The IL1RAP coreceptor similarly possesses extracellular immunoglobulin domains and a cytoplasmic TIR domain, and the assembly of a functional signaling complex requires that both the IL-36 agonist and IL-36R and IL1RAP be simultaneously present and appropriately oriented at the plasma membrane.[8][39]
The structural requirements for IL-36 agonist binding to IL-36R have been systematically investigated through biochemical and structural modeling studies. Full-length recombinant IL-36 receptor ectodomain (the D1-D3 domains) was required for efficient binding of IL-36 cytokines; remarkably, no detectable binding of IL-36 cytokines to a truncated receptor construct containing only domains D1-D2 was observed even at very high (50 micromolar) agonist concentrations, indicating that the D3 domain makes essential contributions to agonist recognition and binding.[5] These findings suggest that the IL-36 recognition surface is distributed across multiple domains of the IL-36R ectodomain and that productive agonist binding requires the full structural context of the complete extracellular portion of the receptor. The specificity of IL-36Ra binding to IL-36R has been confirmed through multiple experimental approaches; when IL-36Ra was tested against IL-1R1 (the receptor for IL-1α and IL-1β), no antagonism of IL-1R1 signaling was observed even at saturating concentrations, demonstrating that IL-36Ra exhibits extraordinary selectivity for IL-36R over the highly homologous IL-1R1.[1][5]
Upon IL-36 agonist binding, the conformational change induced in the IL-36R ectodomain promotes the recruitment and stabilization of IL1RAP at the plasma membrane, leading to formation of the signaling-competent ternary complex.[8][39] This ternary complex brings the two TIR domains of IL-36R and IL1RAP into close proximity and appropriate orientation at the cytoplasmic face of the plasma membrane, enabling the recruitment of the adaptor protein myeloid differentiation protein 88 (MyD88), which interacts with the TIR domains through homotypic TIR-TIR domain interactions.[38][41] The MyD88 adaptor protein subsequently recruits IRAK4 (IL-1 receptor-associated kinase 4) through interaction between the C-terminal region of MyD88 and the death domain of IRAK4, and this interaction initiates a cascade of protein kinase phosphorylation events.[38][41] IRAK4 undergoes autophosphorylation and trans-phosphorylation of IRAK1, leading to activation of IRAK1 kinase activity; subsequently, the activated IRAK proteins phosphorylate TNF receptor-associated factor 6 (TRAF6), an E3 ubiquitin ligase that catalyzes the synthesis of K63-linked polyubiquitin chains on itself and scaffold proteins including the IKK complex components NEMO and RIP1.[41][57]
The K63-linked polyubiquitination of IKK complex components serves as a critical signal for the recruitment and activation of the TGF-β-activated kinase 1 (TAK1) complex, which includes the adapter proteins TAB2 and TAB3 that recognize K63-linked ubiquitin chains through their NZF (zinc-finger) domains.[41][57] TAK1 activation leads to phosphorylation and activation of the inhibitor of κB kinase (IKK) complex, which consists of IKKα, IKKβ, and the structural component NEMO.[41][57] The activated IKK complex phosphorylates the inhibitor of NF-κB (IκBα) protein at specific serine residues, causing IκBα to dissociate from the NF-κB p65/p50 heterodimer and undergo proteasomal degradation, thereby liberating NF-κB for nuclear translocation and binding to κB sites in the promoters of pro-inflammatory genes.[15][41] Simultaneously, the activated MAPK pathway kinases including extracellular signal-regulated kinases (ERK1/2), p38 MAPK, and c-Jun N-terminal kinase (JNK) undergo phosphorylation and activation downstream of TRAF6, leading to phosphorylation of downstream transcription factors including c-Jun and c-Fos.[14][57] The convergent activation of both the NF-κB and MAPK signaling cascades downstream of IL-36R engagement drives rapid and robust transcriptional upregulation of genes encoding pro-inflammatory cytokines, chemokines, adhesion molecules, and antimicrobial peptides.[10][14][20][57]
In the specific context of epithelial cells such as keratinocytes, IL-36 stimulation induces substantial upregulation of gene expression for the chemokine IL-8 (CXCL8), which is potently chemotactic for neutrophils and serves to amplify the inflammatory response by recruiting additional inflammatory cells to the site of IL-36 production.[18] Additional chemokines induced downstream of IL-36R signaling include CCL20 and CXCL1, which also participate in immune cell recruitment, and the expression of genes encoding antimicrobial peptides including LL-37 and human β-defensin-2 (HBD-2) is substantially upregulated in response to IL-36 stimulation.[10][14][20][57] The IL-36-stimulated upregulation of antimicrobial peptide production reflects the evolutionary origin of the IL-36 system as a component of barrier immunity, wherein IL-36 functions to alert and activate epithelial cells and immune cells to the presence of microbial pathogens and to mobilize local innate immune defenses. Furthermore, IL-36 signaling in keratinocytes induces the expression of additional IL-36 agonist cytokines, creating an autocrine amplification loop wherein IL-36-stimulated keratinocytes produce additional IL-36 agonists that further activate surrounding cells, resulting in rapid and substantial amplification of the inflammatory cascade.[10][18] The expression of IL36RN encoding IL-36Ra is also responsive to IL-36 stimulation, creating a negative feedback regulatory mechanism wherein the inflammatory stimulus simultaneously triggers production of the antagonist that limits signal transduction, a regulatory strategy observed for multiple components of the IL-36 signaling system.
Genetic Variation, Mutation Spectrum, and Disease Association
The IL36RN gene is the target of recurrent loss-of-function mutations that account for a substantial proportion of cases of generalized pustular psoriasis (GPP), a potentially life-threatening autoinflammatory skin disorder characterized by widespread pustulation, erythema, and systemic inflammation.[7][21][24][43][50] The clinical significance of IL36RN mutations has been emphasized by the definition of a new disease entity termed DITRA (deficiency of IL-36 receptor antagonist), which encompasses the spectrum of pustular diseases caused by genetic deficiency or impaired function of IL-36Ra.[7][21][50] The frequency of IL36RN mutations in patients with GPP varies substantially depending on geographic location and ethnicity, with reports indicating monoallelic mutations in up to 33.3 percent of GPP patients and biallelic mutations in up to 73.2 percent of GPP patients across various populations, whereas mutation frequencies in patients with plaque psoriasis remain minimal (zero to 11.9 percent for monoallelic mutations) or absent (zero percent for biallelic mutations).[21] These differences in mutation frequency across disease phenotypes provide compelling evidence that IL36RN mutations cause a distinct genetic form of pustular disease that is etiologically separate from common plaque psoriasis.
More than twenty distinct IL36RN gene mutations have been identified in association with various forms of pustular psoriasis and other IL36RN-associated inflammatory conditions.[7][24][43] The mutations identified in IL36RN can be functionally categorized into two major groups: null mutations that result in either complete absence of IL-36Ra protein expression or production of truncated protein fragments that are rapidly degraded, and hypomorphic mutations that permit production of full-length IL-36Ra protein but with impaired biological function, typically manifesting as either reduced antagonistic potency or decreased protein stability.[50] The null mutations identified in IL36RN include point mutations introducing premature stop codons (nonsense mutations) such as c.28C>T (p.Arg10X), c.41C>A (p.Ser14X), and c.280G>T (p.Glu94X), as well as frameshift mutations resulting from insertions or deletions such as c.420_426del (p.Gly141MetfsX29).[50] Additionally, null mutations include missense mutations that severely destabilize the protein, such as c.80T>C (p.Leu27Pro), c.227C>T (p.Pro76Leu), c.368C>T (p.Thr123Met), and c.368C>G (p.Thr123Arg), which result in marked reduction of protein accumulation compared to wild-type IL-36Ra when expressed in cultured cells.[29][50]
The hypomorphic mutations identified in IL36RN include missense mutations that preserve protein expression at near-wild-type levels but impair biological function, such as c.95A>G (p.His32Arg), c.142C>T (p.Arg48Trp), and c.304C>T (p.Arg102Trp), as well as other mutations like c.308C>T (p.Ser113Leu) that show either partial reduction in protein expression or diminished antagonistic activity without complete loss of function.[29][50] Functional studies comparing the antagonistic capacity of different IL36RN mutant proteins have revealed that IL-36Ra variants carrying null mutations exhibit complete functional impairment and fail to repress IL-36-dependent NF-κB activation in reporter assays, whereas hypomorphic mutations show either partial or minimal impairment of antagonistic function.[29][50] These functional distinctions between null and hypomorphic mutations demonstrate important genotype-phenotype correlations within the IL36RN mutation spectrum.
Particular ethnic and geographic variations in IL36RN mutation frequencies and specific mutation types have been documented. The mutation c.115+6T>C, which affects a splice site and generates aberrant splicing of IL36RN transcripts, represents the most frequently observed IL36RN mutation in Asian populations with pustular psoriasis, yet this specific mutation has not been reported in European or African populations.[24][47] In contrast, mutations such as the missense mutation c.338C>T (p.Ser113Leu) are identified as founder mutations in European populations.[26] This geographic variation in mutation spectrum reflects the evolutionary history and population genetics of different human populations and has been proposed to result from the action of founder effects or specific mutational hotspots that differ across populations. For example, the c.368C mutation has been identified as a potential mutational hotspot occurring at a CpG dinucleotide sequence, with this specific nucleotide change identified independently in both European and Japanese patients, suggesting that this particular dinucleotide position exhibits elevated mutability across different populations.[26]
The IL36RN mutation-associated diseases encompass a spectrum of clinical phenotypes that correlate with the functional severity of the underlying mutations. Null mutations causing complete absence of IL-36Ra protein expression show strong associations with the most severe and generalized forms of pustular disease, particularly generalized pustular psoriasis (GPP) and acute generalized exanthematous pustulosis (AGEP).[50][53] In contrast, hypomorphic mutations permitting production of partially functional IL-36Ra are more commonly identified in patients with localized forms of pustular disease, including palmoplantar pustular psoriasis (PPPP), characterized by pustular lesions restricted to the palms and soles, and acrodermatitis continua of Hallopeau (ACH), characterized by progressive pustulation and destructive nail changes at the fingertips and toes.[50][53] This genotype-phenotype correlation suggests that the magnitude of IL-36 dysregulation, which correlates with the degree of functional impairment of IL-36Ra, determines whether disease manifestations are limited to specific anatomical sites or progress to systemic pustulation and systemic inflammation.[50][53]
Beyond the aforementioned major pustular phenotypes, IL36RN mutations have been identified in association with geographic tongue (GT), a benign migratory glossitis characterized by erythematous patches with white borders on the dorsal and lateral surfaces of the tongue that wax and wane over time.[44][45] Investigation of geographic tongue cases revealed that certain IL36RN mutations, particularly the c.115+6T>C splice site mutation, are enriched in patients with geographic tongue, and furthermore, that the presence of IL36RN mutations in geographic tongue patients correlates with the simultaneous presence of generalized pustular psoriasis phenotypes.[44][45] Patients carrying heterozygous IL36RN mutations show reduced but not absent IL-36Ra expression and disproportionately develop tongue lesions rather than generalized skin disease, suggesting that tongue tissue may require higher baseline levels of IL-36Ra antagonistic activity than skin to maintain homeostasis, and that heterozygous deficiency sufficient to disturb tongue immune tolerance may be insufficient to precipitate systemic disease.[44][45] Recent evidence has also linked IL36RN mutations to intestinal inflammation in the context of Crohn's disease, indicating that IL36RN-associated dysregulation of IL-36 signaling may predispose patients to intestinal inflammatory conditions in addition to the historically recognized skin manifestations.[17]
Structural Determinants of IL-36Ra Protein Function and Stability
Recent investigations employing integrated in silico and in vitro approaches have systematically identified the amino acid residues within the IL-36Ra protein structure that are critical for maintaining protein folding stability versus those that are essential for the molecular recognition of and binding to the IL-36 receptor.[29][49] Saturation mutagenesis studies modeling the effects of all possible amino acid substitutions at each position within the IL-36Ra protein identified a set of particularly intolerant residues that are predicted to be critical for protein stability, with the most critical residues demonstrating a marked preference for localization within β-strands of the protein core rather than on the protein surface.[29][49] These computationally predicted critical residues demonstrate strong evolutionary constraint, showing significantly elevated genomic evolutionary rate profiling (GERP) scores relative to residues where amino acid substitutions are more commonly tolerated.[29][49] Three spatially adjacent clusters of these critical residues were identified spanning amino acids 56-61, 96-104, and 119-123, with amino acids at these positions forming the hydrophobic core of the protein and exhibiting high conservation across different IL-1 family members including IL-1Ra, IL-36α, IL-36β, IL-36γ, and IL-38, suggesting the β-trefoil fold structure represents an evolutionarily optimized protein architecture for IL-1 family cytokines.[29][49]
Functional characterization of IL36RN missense mutations identified in GPP patients reveals that mutations affecting the protein core and destabilizing the β-trefoil fold structure result in marked reductions in steady-state protein accumulation and substantial loss of antagonistic function, whereas surface-located residues represent binding determinants whose disruption may impair IL-36Ra/IL-36R interaction while preserving protein stability.[29][49] Notably, experimental validation of computationally predicted destabilizing mutations confirmed that several GPP-associated alleles including p.Pro27Leu, p.Ser113Leu, p.Ile42Asn, p.Glu112Lys, and p.Thr123Arg resulted in marked reductions of IL-36Ra protein accumulation exceeding four-fold when expressed in cultured cells, whereas most population-frequency variants (identified in unaffected individuals) did not substantially destabilize the protein.[29] These findings establish a clear distinction between disease-associated and population-tolerated variation within the IL36RN gene, wherein disease-associated mutations predominantly disrupt critical protein-folding determinants whereas population variants tend to represent tolerated changes in surface-exposed regions of the protein.
The binding interface between IL-36Ra and IL-36R involves specific hydrophilic amino acid residues on the IL-36Ra surface that make critical contacts with complementary residues within the IL-36R ectodomain. In silico saturation scanning using machine learning algorithms predicting the effects of amino acid substitutions on protein binding affinity (specifically the mCSM and mCSM-PPI2 tools) identified several key IL-36Ra residues whose mutation was predicted to substantially reduce IL-36Ra/IL-36R binding affinity.[29][49] Significantly, the predicted impact of IL36RN mutations on IL-36Ra/IL-36R binding affinity as quantified by changes in Gibbs free energy (ΔΔG) demonstrated significant correlation with experimental measurements of IL-36 signaling in cell-based assays measuring IL-8 production downstream of IL-36R engagement.[29] This finding suggests that surface-accessible residues and their specific interactions with the IL-36R ectodomain represent distinct structural determinants of antagonistic function separate from the core residues that determine protein stability, and that the cumulative effect of IL36RN mutations on either stability or binding affinity drives the loss of antagonistic function observed in patients.[29]
Expression Regulation and Feedback Control Mechanisms
The expression of IL36RN is dynamically regulated in response to inflammatory stimuli and represents an important component of feedback regulatory mechanisms that limit the magnitude and duration of IL-36-driven inflammatory responses. IL-36 stimulation of keratinocytes results in substantial upregulation of IL36RN mRNA expression through MyD88-dependent mechanisms, indicating that IL-36 agonist stimulation triggers transcriptional induction of the antagonist gene, creating a classical negative feedback regulatory circuit.[16] This negative feedback architecture reflects an evolutionary design principle wherein the primary inflammatory signal (IL-36 agonist production) simultaneously triggers the production of the antagonist (IL-36Ra) that will ultimately limit signal transduction, thereby preventing excessive or uncontrolled inflammatory amplification. The expression of IL36RN is also upregulated in response to other pro-inflammatory signals including stimulation with TNF-α, IFN-γ, and IL-17A, inflammatory mediators that are known to contribute to psoriatic inflammation and that participate in crosstalk with the IL-36 signaling pathway.[35] In the context of systemic treatment of psoriasis with anti-IL-17A monoclonal antibodies such as secukinumab, clinical improvement of disease correlates with upregulation of serum IL-36Ra levels, providing clinical evidence for the importance of IL-36Ra expression in suppressing psoriatic inflammation and suggesting that IL-17A suppression permits compensatory upregulation of IL-36Ra that contributes to disease amelioration.[59]
Beyond keratinocyte-derived IL-36Ra, the IL-36 signaling system appears to involve production of a soluble IL-36 receptor (sIL-36R) that represents an alternative spliced transcript of the IL1RL2 gene encoding the transmembrane IL-36 receptor.[57] This soluble receptor, containing the extracellular domain of the IL-36 receptor but lacking the transmembrane and intracellular signaling domains, can bind IL-36 agonists in a dose-dependent manner and compete with the membrane-bound IL-36R for agonist binding, thereby suppressing IL-36 signaling through a mechanism distinct from IL-36Ra competitive antagonism.[57] The production of sIL-36R is regulated by alternative splicing of IL1RL2 mRNA, a process mediated by the RNA helicase DDX5, and interestingly, IL-17D signaling suppresses DDX5 expression, providing a mechanistic link between IL-17D signaling and reduced sIL-36R generation and thereby permitting enhanced IL-36 signaling during contexts of active IL-17 production.[57] This complex regulatory architecture involving cooperative production of IL-36Ra antagonist and soluble receptor suggests that IL-36 biology represents an extensively evolved system with multiple independent regulatory mechanisms that collectively serve to fine-tune the amplitude and kinetics of IL-36-driven inflammatory responses.
Therapeutic Applications and Recent Clinical Developments
The critical role of IL-36Ra deficiency in driving the pathogenesis of generalized pustular psoriasis has prompted the development of therapeutic strategies specifically targeting the IL-36 signaling pathway. The discovery of IL36RN mutations as a major cause of GPP and the clinical validation of this genetic basis through animal models have provided strong justification for IL-36R-targeted therapeutics as a treatment strategy for GPP and related conditions. In 2019, spesolimab (also known as BI 655130 and marketed as Spevigo), a humanized monoclonal antibody that specifically binds to the human IL-36R and blocks IL-36 agonist engagement and downstream signaling, was developed and proceeded to clinical trial evaluation for GPP treatment.[51][54] Phase two clinical trials of intravenous spesolimab administered as a single infusion in the context of acute GPP flares demonstrated substantial clinical efficacy, with fifty-four percent of spesolimab-treated patients achieving a pustulation subscore of zero on the GPP Physician Global Assessment (GPPGA) scale by day eight post-infusion compared to only six percent of placebo-treated patients, and forty-three percent of spesolimab recipients achieving a complete resolution of GPPGA total score (zero) compared to eleven percent of placebo recipients.[51] Additional phase two trials of subcutaneous spesolimab administered for maintenance therapy of GPP demonstrated dose-dependent efficacy in preventing subsequent GPP flares, with twenty-three percent of patients receiving low-dose spesolimab experiencing flares by week forty-eight compared to twenty-nine percent receiving medium-dose and fifty-two percent receiving placebo, corresponding to hazard ratios for time to flare of 0.16, 0.35, and 0.47 respectively relative to placebo.[51]
The safety profile of spesolimab has been favorable across clinical trials, with infection rates and adverse event frequencies similar to placebo-treated controls, indicating that transient IL-36R blockade does not substantially compromise immune defense mechanisms.[51] However, additional investigation has revealed that spesolimab treatment mitigates residual skin inflammation that persists even after resolution of acute pustular flares, suggesting that complete suppression of IL-36R signaling may produce benefits beyond immediate pustule resolution and may promote restoration of skin barrier function and normalization of immune homeostasis.[54] The clinical efficacy of spesolimab against acute GPP flares and in preventing recurrent flares has led to regulatory approval for this therapeutic agent in multiple countries, representing the first IL-36-targeted monoclonal antibody approved for clinical use and establishing a new therapeutic paradigm for the treatment of IL36RN-associated pustular disease.[51]
In addition to monoclonal antibody-based approaches targeting IL-36R directly, alternative therapeutic strategies targeting the IL-36 signaling pathway have been explored. Small-molecule inhibitors capable of selectively blocking IL-36R signaling have been developed, with recent structures demonstrating that peptide inhibitors can be designed as orthosteric inhibitors that compete with IL-36 agonists for IL-36R binding.[5] These peptide inhibitors demonstrate potent dose-dependent inhibition of all three IL-36 agonists (IL-36α, IL-36β, and IL-36γ) in reporter assays measuring IL-36R signaling in recombinant cell lines overexpressing the receptor, and importantly, these peptide inhibitors exhibit specificity for IL-36R over IL-1R1, avoiding off-target effects on IL-1 signaling that might compromise immune defense.[5] Human skin explant studies employing tissue from healthy donors stimulated with IL-36α demonstrated that peptide IL-36R inhibitors suppress multiple IL-36-responsive genes including IL36RN itself, IL-23, and the antimicrobial peptide gene SERPINB6 in a dose-dependent manner, though higher concentrations of the peptide were required for gene suppression in this more physiologically relevant tissue system compared to cultured cells, possibly reflecting the effects of plasma protein binding and tissue penetration.[5]
Beyond direct IL-36R antagonism, alternative approaches targeting the IL-36 signaling pathway have been explored, including targeting the IL-1 receptor accessory protein (IL1RAP) that functions as the obligate coreceptor for IL-36R signaling. A monoclonal antibody targeting IL1RAP (CAN10) has entered clinical trials for systemic sclerosis and fibrotic diseases, with the hypothesis that blocking IL1RAP would simultaneously prevent signaling downstream of IL-1, IL-33, and IL-36, thereby blocking multiple profibrotic cytokine pathways simultaneously.[11][42] This approach represents a broader therapeutic strategy that exploits the shared usage of IL1RAP as a coreceptor by multiple IL-1 family cytokines to achieve broader anti-inflammatory effects compared to targeting a single agonist or receptor.[11][42] Additionally, transcriptional control approaches designed to upregulate IL36RN expression or inhibit the production of IL-36 agonists represent potential therapeutic strategies, though these approaches remain largely in preclinical development stages.
Conclusion
The IL36RN gene encodes the interleukin-36 receptor antagonist (IL-36Ra), a critical negative regulator of epithelial barrier inflammation that functions as a competitive antagonist of the pro-inflammatory IL-36 agonist cytokines through high-affinity binding to the IL-36 receptor and prevention of the recruitment of the essential IL1RAP coreceptor required for signal transduction. The structural architecture of IL-36Ra reflects evolutionary conservation of the β-trefoil fold shared among IL-1 family members, with specific amino acid residues concentrated in the protein core that determine protein stability and surface-exposed residues that mediate receptor recognition and binding. Loss-of-function mutations in IL36RN represent a major cause of generalized pustular psoriasis and have been unified into the disease entity DITRA (deficiency of IL-36 receptor antagonist), a distinct autoinflammatory condition caused by uncontrolled IL-36 signaling at epithelial barriers. The phenotypic manifestations of IL36RN deficiency range from localized pustular disease associated with hypomorphic mutations to severe systemic pustulation and systemic inflammation associated with null mutations, reflecting the extent of IL-36 dysregulation. Recent therapeutic advances including the clinical approval of spesolimab, a monoclonal antibody blocking IL-36R, have validated IL-36R as a tractable drug target and have established effective treatment strategies for previously intractable IL36RN-associated pustular diseases. Ongoing investigation of IL-36 biology continues to reveal additional regulatory mechanisms including the production of soluble IL-36 receptor variants and the context-dependent engagement of alternative receptors including SIGIRR, suggesting that this signaling system possesses multiple independent regulatory circuits that collectively orchestrate epithelial barrier immune homeostasis. Future therapeutic development may exploit these multiple regulatory mechanisms to achieve enhanced efficacy in treating not only pustular psoriasis but also emerging evidence for IL-36 dysregulation in other inflammatory diseases including Crohn's disease and other autoinflammatory conditions. The systematic molecular characterization of IL36RN mutations and their functional consequences has provided unprecedented insights into the structure-function relationships within the IL-1 family and has established a paradigm wherein genetic deficiency of a negative regulator drives specific inflammatory disease phenotypes, highlighting the essential biological role of this antagonist in maintaining immune tolerance at epithelial barriers and preventing excessive inflammatory activation in response to commensal microbes and environmental stimuli.
Citations
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=IL36RN
- https://en.wikipedia.org/wiki/Interleukin_36_receptor_antagonist
- https://www.uniprot.org/uniprotkb/Q9UBH0/entry
- https://www.ncbi.nlm.nih.gov/gene/26525
- https://www.nature.com/articles/s41467-025-56601-7
- https://www.uniprot.org/uniprotkb/Q9UBH0/history
- https://medlineplus.gov/genetics/gene/il36rn/
- https://reactome.org/content/detail/R-HSA-8848314
- https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2019.01162/full
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6839525/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC11420747/
- https://www.science.org/doi/10.1126/sciimmunol.aax1686
- https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2019.00317/full
- https://www.nature.com/articles/s41392-020-00312-6
- https://www.uniprot.org/uniprotkb/Q9QYY1/entry
- https://www.embopress.org/doi/10.1038/s44321-025-00245-z
- https://pmc.ncbi.nlm.nih.gov/articles/PMC7043067/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC10988704/
- https://onlinelibrary.wiley.com/doi/10.1111/jdv.19935
- https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2022.1035037/full
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6904269/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC4762540/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC4821354/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC7035875/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC10824670/
- https://febs.onlinelibrary.wiley.com/doi/abs/10.1002/2211-5463.12044
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8054633/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC7190273/
- https://pubmed.ncbi.nlm.nih.gov/33443029/
- https://insight.jci.org/articles/view/123390
- https://pmc.ncbi.nlm.nih.gov/articles/PMC9020223/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC9972678/
- https://onlinelibrary.wiley.com/doi/10.1111/1346-8138.12227
- https://pubmed.ncbi.nlm.nih.gov/27900482/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5258799/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC11964088/
- https://www.actasdermo.org/en-geographic-tongue-what-dermatologist-should-articulo-S1578219019301489
- https://pubmed.ncbi.nlm.nih.gov/27220475/
- https://pubmed.ncbi.nlm.nih.gov/38755971/
- https://www.medicaljournals.se/acta/content/html/10.2340/00015555-3388
- https://pubmed.ncbi.nlm.nih.gov/40591420/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC4298222/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6650959/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC10679229/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC12442919/
- https://www.nature.com/articles/s41419-018-1143-3
- https://bibliographie.uni-tuebingen.de/xmlui/bitstream/handle/10900/109643/dissertation_Anne_M%C3%BCller_bib.pdf?sequence=5