Deep Research
Falcon
(PHYKPL-deep-research-falcon.md)
Falcon
(PHYKPL-deep-research-falcon.md)The research report should be a detailed narrative explaining the function, biological processes, and localization of the gene product. Citations should be given for all claims.
You should prioritize authoritative reviews and primary scientific literature when conducting research. You can supplement
this with annotations you find in gene/protein databases, but these can be outdated or inaccurate.
We are specifically interested in the primary function of the gene - for enzymes, what reaction is catalyzed, and what is the substrate specificity? For transporters, what is the substrate? For structural proteins or adapters, what is the broader structural role? For signaling molecules, what is the role in the pathway.
We are interested in where in or outside the cell the gene product carries out its function.
We are also interested in the signaling or biochemical pathways in which the gene functions. We are less interested in broad pleiotropic effects, except where these elucidate the precise role.
Include evidence where possible. We are interested in both experimental evidence as well as inference from structure, evolution, or bioinformatic analysis. Precise studies should be prioritized over high-throughput, where available.
Research report: Human PHYKPL (AGXT2L2; UniProt Q8IUZ5)
Executive summary
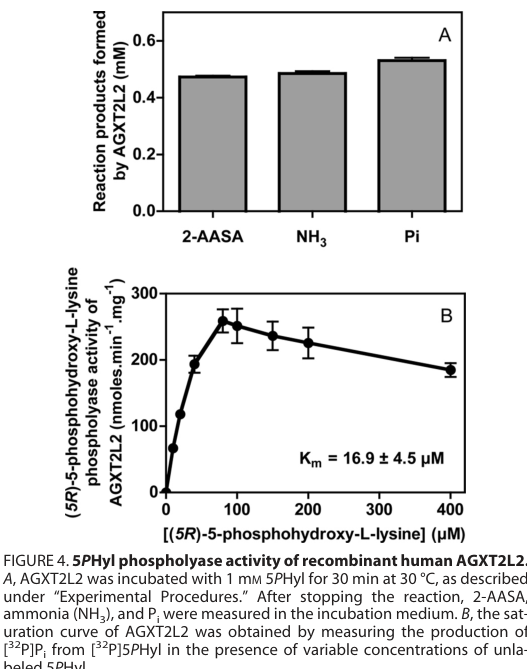
PHYKPL (synonym AGXT2L2) encodes a pyridoxal-5′-phosphate (PLP)-dependent ammoniophospholyase whose primary, experimentally demonstrated function is to catalyze elimination of 5-phosphohydroxy-L-lysine (5PHyl) to yield 2-aminoadipate semialdehyde (2-AASA), inorganic phosphate (Pi), and ammonia (NH3). This reaction places PHYKPL in a defined pathway for hydroxylysine catabolism downstream of a 5-hydroxylysine kinase (AGPHD1/HYKK). The best-supported Mendelian phenotype link is phosphohydroxylysinuria, consistent with expectation that loss of PHYKPL activity increases urinary 5PHyl. Recent (2023–2024) literature retrieved here contains only indirect mentions (e.g., homology-based discussion in PLP enzyme/heme metabolism contexts), while disease-association resources (Open Targets) continue to track a strong association to phosphohydroxylysinuria and weaker GWAS-derived associations to several complex diseases. (veigadacunha2012molecularidentificationof pages 6-7, veigadacunha2012molecularidentificationof pages 1-2, OpenTargets Search: -PHYKPL,AGXT2L2, key2024clppnulleukaryoteswitha pages 2-4)
1) Key concepts and definitions (current understanding)
1.1 Gene/protein identity and nomenclature
The UniProt entry Q8IUZ5 corresponds to human PHYKPL, also known as AGXT2L2, annotated as 5-phosphohydroxy-L-lysine phospho-lyase (EC 4.2.3.134). Primary biochemistry establishing this identity comes from recombinant expression and activity assays showing specific cleavage of 5PHyl to Pi, NH3 and 2-AASA. (veigadacunha2012molecularidentificationof pages 1-2, veigadacunha2012molecularidentificationof pages 3-4)
1.2 Enzyme class: PLP-dependent “ammoniophospholyase”
Although PHYKPL/AGXT2L2 is homologous to class-III PLP-dependent aminotransferases, the experimentally supported activity is a PLP-dependent elimination (phospho-lyase) rather than transamination. In the defining biochemical study, AGXT2L2 was purified as a yellow PLP-binding protein, consistent with PLP cofactor association, and aminotransferase activity was not detected in the authors’ assays for the AGXT2L1/2 paralogs. (veigadacunha2012molecularidentificationof pages 3-4, veigadacunha2012molecularidentificationof pages 7-8)
1.3 Reaction and pathway placement: hydroxylysine catabolism
PHYKPL catalyzes:
5-phosphohydroxy-L-lysine (5PHyl) → 2-aminoadipate semialdehyde (2-AASA) + Pi + NH3
This reaction functions downstream of a kinase (AGPHD1) that phosphorylates 5-hydroxy-L-lysine to create the 5PHyl substrate, establishing a two-step metabolic route for hydroxylysine degradation. (veigadacunha2012molecularidentificationof pages 1-2, veigadacunha2012molecularidentificationof pages 5-6)
2) Functional biochemistry: substrate specificity and kinetics
2.1 Experimental evidence for substrate specificity
The key functional assignment is supported by direct in vitro assays using recombinant human AGXT2L2, where production of Pi, NH3, and the aldehyde product 2-AASA (measured as a derivatized product by RP-HPLC) was observed after incubation with 5PHyl. (veigadacunha2012molecularidentificationof pages 3-4)
The defining study also reports that, under their conditions, the AGXT2L1/2 paralogs did not show detectable aminotransferase activity in the specific assays performed, reinforcing that the physiologically relevant activity for AGXT2L2 is the phospho-lyase reaction. (veigadacunha2012molecularidentificationof pages 7-8)
2.2 Quantitative kinetics and inhibition
Kinetic parameters reported for recombinant human AGXT2L2 acting on 5PHyl include:
- Km(5PHyl) = 16.9 ± 4.5 μM
- Vmax = 256 ± 15 nmol·min⁻¹·mg⁻¹ protein
The saturation behavior shows substrate inhibition at higher 5PHyl concentrations (notably above ~100 μM in the saturation curve) and inhibition by inorganic phosphate, with the highest tested Pi reducing activity by ~50% in the reported ammonia-production readout. (veigadacunha2012molecularidentificationof pages 6-7, veigadacunha2012molecularidentificationof media 1ef79764)
3) Cellular/subcellular localization and expression
3.1 Subcellular localization
AGXT2L2 (PHYKPL) is described as a cytosolic enzyme, consistent with TargetP and Psort II predictions cited in the biochemical characterization paper. (veigadacunha2012molecularidentificationof pages 6-7)
3.2 Tissue expression and physiological context
Direct quantitative tissue expression profiling for PHYKPL/AGXT2L2 was not available in the retrieved full-text excerpts. However:
- The AGXT2L2 coding sequence was cloned from human kidney cDNA, consistent with kidney expression. (veigadacunha2012molecularidentificationof pages 2-3)
- The same study notes that 5PHyl is a normal constituent of adult brain, which motivates concern that loss of AGXT2L2 could cause brain accumulation of 5PHyl with neurological effects (see disease section). (veigadacunha2012molecularidentificationof pages 6-7)
4) Disease associations, applications, and real-world implementations
4.1 Mendelian/metabolic disease: phosphohydroxylysinuria
The defining 2012 biochemical study predicts that mutations in AGXT2L2 should result in increased urinary 5PHyl excretion, and notes that only two unrelated patients with phosphohydroxylysinuria and neurological abnormalities had been reported in earlier literature at that time. (veigadacunha2012molecularidentificationof pages 6-7)
From an implementation perspective, this implies a clinically actionable diagnostic logic: urinary 5PHyl elevation is a plausible biochemical marker indicating deficiency in the AGPHD1→AGXT2L2 hydroxylysine-catabolic pathway, and the molecular identification of AGXT2L2 enables confirmatory genetic diagnosis. (veigadacunha2012molecularidentificationof pages 6-7, veigadacunha2012molecularidentificationof pages 1-2)
4.2 Complex disease genetics (database-level evidence)
Open Targets reports PHYKPL associations including:
- Phosphohydroxylysinuria (highest score among listed diseases; composite score ~0.569)
- GWAS-derived genetic associations to ulcerative colitis, dental caries, glomerulonephritis, and primary thrombocytopenia (scores roughly ~0.27–0.32 in the displayed output)
These complex-disease links are primarily genetic association signals rather than established mechanistic disease roles for the enzyme. (OpenTargets Search: -PHYKPL,AGXT2L2)
4.3 Clinical trials and therapeutics
A clinical-trials search for “PHYKPL OR AGXT2L2” did not yield relevant interventional trials targeting this gene/protein in the retrieved results. (OpenTargets Search: -PHYKPL,AGXT2L2)
5) Recent developments and latest research (prioritizing 2023–2024)
5.1 2024: broader PLP-enzyme/mitochondrial metabolism discussion
A 2024 study focused on CLPP/CLPX biology and metabolomics mentions PHYKPL as one of several PLP-dependent proteins with sequence homology to bacterial HemL (glutamate-1-semialdehyde aminomutase), in the context of hypothesized alternative δ-aminolevulinic acid (δALA) production routes. This is not new experimental evidence about PHYKPL’s physiological function, but illustrates how PHYKPL appears in comparative discussions of PLP-enzyme repertoires. (key2024clppnulleukaryoteswitha pages 2-4)
5.2 2023–2024 gaps in the retrievable corpus
Within the documents retrievable here, there were no 2023–2024 primary studies that provided new direct biochemical characterization, localization by microscopy/proteomics, or targeted functional genetics for human PHYKPL beyond the established 2012 biochemistry and database association updates. Therefore, current best practice for functional annotation remains anchored in the 2012 enzymology while monitoring genetic databases and emerging omics studies for new physiology or disease insights. (key2024clppnulleukaryoteswitha pages 2-4, OpenTargets Search: -PHYKPL,AGXT2L2)
6) Expert interpretation and analysis (authoritative sources)
The most authoritative mechanistic evidence is the enzymology work establishing that vertebrate genomes encode AGXT2L1/AGXT2L2 as enzymes acting on phosphorylated terminal amines, and that AGXT2L2 specifically is the 5PHyl phospho-lyase. The authors explicitly connect this to explain rare neurometabolic phenotypes, and they propose AGPHD1 and AGXT2L2 as likely mutated enzymes in 5-hydroxylysinuria and phosphohydroxylysinuria. (veigadacunha2012molecularidentificationof pages 1-2)
A key interpretive nuance is that phylogenetic homology to aminotransferases led earlier studies to test AGXT2L2 as a transaminase; e.g., a 2009 recombinant expression study screened AGXT2L2 for glyoxylate transamination but did not find glycine production under their conditions, and noted that reaction preferences were still unclear at that time. The subsequent 2012 work resolved the principal activity as a phospho-lyase, illustrating how enzyme superfamily membership can mislead functional inference without substrate-guided biochemistry. (donini2009recombinantproductionof pages 10-12, veigadacunha2012molecularidentificationof pages 1-2)
7) Key statistics and data points from studies
- Enzyme kinetics (recombinant human PHYKPL/AGXT2L2): Km(5PHyl) 16.9 ± 4.5 μM; Vmax 256 ± 15 nmol·min⁻¹·mg⁻¹; substrate inhibition above ~100 μM 5PHyl. (veigadacunha2012molecularidentificationof media 1ef79764, veigadacunha2012molecularidentificationof pages 6-7)
- Disease case count noted in foundational report: phosphohydroxylysinuria had been reported in two unrelated patients with neurological abnormalities (as summarized in the 2012 study). (veigadacunha2012molecularidentificationof pages 6-7)
- Database-level disease association scores (Open Targets): phosphohydroxylysinuria score ~0.569; ulcerative colitis ~0.319; dental caries ~0.310; glomerulonephritis ~0.296; primary thrombocytopenia ~0.269 (as displayed). (OpenTargets Search: -PHYKPL,AGXT2L2)
Evidence map (summary table)
| Aspect | Finding | Key evidence/source (paper and year) | Notes/limitations |
|---|---|---|---|
| Gene/protein identity | Human PHYKPL / AGXT2L2 encodes 5-phosphohydroxy-L-lysine phospho-lyase; a PLP-dependent enzyme matching UniProt Q8IUZ5 | Veiga-da-Cunha et al., J Biol Chem (2012) (veigadacunha2012molecularidentificationof pages 1-2, veigadacunha2012molecularidentificationof pages 3-4) | Identity is well matched to the UniProt entry; older literature often used the synonym AGXT2L2 rather than PHYKPL |
| Enzymatic reaction | Catalyzes breakdown of 5-phosphohydroxy-L-lysine (5PHyl) to 2-aminoadipate semialdehyde (2-AASA) + NH3 + Pi | Veiga-da-Cunha et al. (2012) (veigadacunha2012molecularidentificationof pages 6-7, veigadacunha2012molecularidentificationof pages 1-2, veigadacunha2012molecularidentificationof pages 3-4) | Experimentally demonstrated with recombinant human enzyme; this is the primary functional annotation |
| Cofactor / enzyme class | Pyridoxal phosphate (PLP)-dependent ammoniophospholyase within the class-III aminotransferase-fold family | Veiga-da-Cunha et al. (2012) (veigadacunha2012molecularidentificationof pages 1-2, veigadacunha2012molecularidentificationof pages 3-4) | Despite homology to aminotransferases, no aminotransferase activity was detected in the reported assays |
| Key kinetics | Km = 16.9 ± 4.5 µM for 5PHyl; Vmax = 256 ± 15 nmol·min⁻¹·mg⁻¹ protein | Veiga-da-Cunha et al. (2012), Figure 4/text (veigadacunha2012molecularidentificationof pages 6-7, veigadacunha2012molecularidentificationof media 1ef79764) | Saturation curve showed substrate inhibition above ~100 µM 5PHyl |
| Product / inhibitor behavior | Reaction yields stoichiometric phosphate, ammonia, and 2-AASA; inorganic phosphate inhibits activity, with ~50% reduction at the highest tested Pi (2–10 mM range tested) | Veiga-da-Cunha et al. (2012) (veigadacunha2012molecularidentificationof pages 6-7) | Supports assignment as a phospho-lyase; inhibition data are biochemical, not in vivo physiology |
| Evidence type | Function established by recombinant human protein expression, purification, and in vitro enzyme assays measuring Pi, NH3, and 2-AASA formation | Veiga-da-Cunha et al. (2012) (veigadacunha2012molecularidentificationof pages 3-4) | Strong primary-biochemistry evidence; human cellular knockout/knockdown studies remain limited in retrieved sources |
| Pathway context | Functions in hydroxylysine catabolism downstream of AGPHD1/HYKK (5-hydroxy-L-lysine kinase), converting phosphorylated hydroxylysine to 2-AASA | Veiga-da-Cunha et al. (2012); Van Schaftingen et al. (2015) (veigadacunha2012molecularidentificationof pages 5-6, veigadacunha2012molecularidentificationof pages 1-2) | Pathway placement is well supported biochemically, but broader physiological flux regulation in humans is still sparsely characterized |
| Localization | Reported/predicted as cytosolic based on bioinformatic localization analyses | Veiga-da-Cunha et al. (2012) (veigadacunha2012molecularidentificationof pages 6-7, veigadacunha2012molecularidentificationof pages 5-6) | Retrieved evidence indicates prediction rather than extensive direct microscopy/proteomics confirmation |
| Disease / Mendelian link | Proposed disease gene for phosphohydroxylysinuria; authors expected AGXT2L2 defects to elevate urinary 5PHyl | Veiga-da-Cunha et al. (2012); Open Targets summary (veigadacunha2012molecularidentificationof pages 6-7, OpenTargets Search: -PHYKPL,AGXT2L2) | A 2013 JIMD mutation paper was identified in search results but not retrievable here; therefore this row relies on the 2012 primary paper plus database-level association evidence |
| Other disease associations | Open Targets lists additional associations with ulcerative colitis, dental caries, glomerulonephritis, and primary thrombocytopenia besides phosphohydroxylysinuria | Open Targets Platform query (accessed via tool context) (OpenTargets Search: -PHYKPL,AGXT2L2) | These are mostly genetic-association/database links, not direct mechanistic validation specific to PHYKPL biology |
| Recent 2023–2024 mentions | 2024 literature mentions PHYKPL mainly as a PLP-enzyme homolog in comparative/heme-metabolism discussion rather than providing new direct functional data | Key et al., Biomolecules (2024) (key2024clppnulleukaryoteswitha pages 2-4) | No major 2023–2024 primary studies with new biochemical or localization data for human PHYKPL were retrieved |
| Current annotation confidence | High confidence for core enzymatic function; moderate-to-low confidence for localization detail, tissue physiology, and broader disease relevance | Synthesis of primary biochemistry and database evidence (veigadacunha2012molecularidentificationof pages 6-7, veigadacunha2012molecularidentificationof pages 1-2, OpenTargets Search: -PHYKPL,AGXT2L2, veigadacunha2012molecularidentificationof media 1ef79764) | Literature on this specific human protein remains limited compared with better-studied metabolic enzymes |
Table: This table summarizes the core functional annotation of human PHYKPL/AGXT2L2, including its experimentally demonstrated phospho-lyase reaction, kinetic parameters, pathway placement, localization, and disease links. It is useful as a compact evidence map separating strong biochemical findings from more tentative disease-association data.
URLs and publication dates (key sources)
- Veiga-da-Cunha M. et al. “Molecular Identification of Hydroxylysine Kinase and of Ammoniophospholyases Acting on 5-Phosphohydroxy-L-lysine and Phosphoethanolamine.” Journal of Biological Chemistry (Mar 2012). https://doi.org/10.1074/jbc.M111.323485 (veigadacunha2012molecularidentificationof pages 1-2, veigadacunha2012molecularidentificationof media 1ef79764)
- Key J. et al. “CLPP-Null Eukaryotes with Excess Heme Biosynthesis Show Reduced L-arginine Levels, Probably via CLPX-Mediated OAT Activation.” Biomolecules (Feb 2024). https://doi.org/10.3390/biom14020241 (key2024clppnulleukaryoteswitha pages 2-4)
- Open Targets Platform disease-target associations for PHYKPL (queried via tool; platform described in Nucleic Acids Research, 2025). https://platform.opentargets.org/ (OpenTargets Search: -PHYKPL,AGXT2L2)
Limitations of this review
A key limitation is that an apparently central Mendelian genetics paper (“Mutations in the AGXT2L2 gene cause phosphohydroxylysinuria,” J Inherit Metab Dis, 2013) was identified by the search system but could not be retrieved in full text in this run, limiting direct quotation of variant types, patient counts, and clinical phenotypes beyond what is summarized in the 2012 biochemical report and Open Targets. (veigadacunha2012molecularidentificationof pages 6-7, OpenTargets Search: -PHYKPL,AGXT2L2)
References
-
(veigadacunha2012molecularidentificationof pages 6-7): Maria Veiga-da-Cunha, Farah Hadi, Thomas Balligand, Vincent Stroobant, and Emile Van Schaftingen. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-l-lysine and phosphoethanolamine. Journal of Biological Chemistry, 287:7246-7255, Mar 2012. URL: https://doi.org/10.1074/jbc.m111.323485, doi:10.1074/jbc.m111.323485. This article has 71 citations and is from a domain leading peer-reviewed journal.
-
(veigadacunha2012molecularidentificationof pages 1-2): Maria Veiga-da-Cunha, Farah Hadi, Thomas Balligand, Vincent Stroobant, and Emile Van Schaftingen. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-l-lysine and phosphoethanolamine. Journal of Biological Chemistry, 287:7246-7255, Mar 2012. URL: https://doi.org/10.1074/jbc.m111.323485, doi:10.1074/jbc.m111.323485. This article has 71 citations and is from a domain leading peer-reviewed journal.
-
(OpenTargets Search: -PHYKPL,AGXT2L2): Open Targets Query (-PHYKPL,AGXT2L2, 5 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(key2024clppnulleukaryoteswitha pages 2-4): Jana Key, Suzana Gispert, Arvind Reddy Kandi, Daniela Heinz, Andrea Hamann, Heinz D. Osiewacz, David Meierhofer, and Georg Auburger. Clpp-null eukaryotes with excess heme biosynthesis show reduced l-arginine levels, probably via clpx-mediated oat activation. Biomolecules, 14:241, Feb 2024. URL: https://doi.org/10.3390/biom14020241, doi:10.3390/biom14020241. This article has 2 citations.
-
(veigadacunha2012molecularidentificationof pages 3-4): Maria Veiga-da-Cunha, Farah Hadi, Thomas Balligand, Vincent Stroobant, and Emile Van Schaftingen. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-l-lysine and phosphoethanolamine. Journal of Biological Chemistry, 287:7246-7255, Mar 2012. URL: https://doi.org/10.1074/jbc.m111.323485, doi:10.1074/jbc.m111.323485. This article has 71 citations and is from a domain leading peer-reviewed journal.
-
(veigadacunha2012molecularidentificationof pages 7-8): Maria Veiga-da-Cunha, Farah Hadi, Thomas Balligand, Vincent Stroobant, and Emile Van Schaftingen. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-l-lysine and phosphoethanolamine. Journal of Biological Chemistry, 287:7246-7255, Mar 2012. URL: https://doi.org/10.1074/jbc.m111.323485, doi:10.1074/jbc.m111.323485. This article has 71 citations and is from a domain leading peer-reviewed journal.
-
(veigadacunha2012molecularidentificationof pages 5-6): Maria Veiga-da-Cunha, Farah Hadi, Thomas Balligand, Vincent Stroobant, and Emile Van Schaftingen. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-l-lysine and phosphoethanolamine. Journal of Biological Chemistry, 287:7246-7255, Mar 2012. URL: https://doi.org/10.1074/jbc.m111.323485, doi:10.1074/jbc.m111.323485. This article has 71 citations and is from a domain leading peer-reviewed journal.
-
(veigadacunha2012molecularidentificationof media 1ef79764): Maria Veiga-da-Cunha, Farah Hadi, Thomas Balligand, Vincent Stroobant, and Emile Van Schaftingen. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-l-lysine and phosphoethanolamine. Journal of Biological Chemistry, 287:7246-7255, Mar 2012. URL: https://doi.org/10.1074/jbc.m111.323485, doi:10.1074/jbc.m111.323485. This article has 71 citations and is from a domain leading peer-reviewed journal.
-
(veigadacunha2012molecularidentificationof pages 2-3): Maria Veiga-da-Cunha, Farah Hadi, Thomas Balligand, Vincent Stroobant, and Emile Van Schaftingen. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-l-lysine and phosphoethanolamine. Journal of Biological Chemistry, 287:7246-7255, Mar 2012. URL: https://doi.org/10.1074/jbc.m111.323485, doi:10.1074/jbc.m111.323485. This article has 71 citations and is from a domain leading peer-reviewed journal.
-
(donini2009recombinantproductionof pages 10-12): Stefano Donini, Manuela Ferrari, Chiara Fedeli, Marco Faini, Ilaria Lamberto, Ada Serena Marletta, Lara Mellini, Michela Panini, Riccardo Percudani, Loredano Pollegioni, Laura Caldinelli, Stefania Petrucco, and Alessio Peracchi. Recombinant production of eight human cytosolic aminotransferases and assessment of their potential involvement in glyoxylate metabolism. The Biochemical journal, 422 2:265-72, Sep 2009. URL: https://doi.org/10.1042/bj20090748, doi:10.1042/bj20090748. This article has 38 citations.
Artifacts
Citations
- veigadacunha2012molecularidentificationof pages 3-4
- veigadacunha2012molecularidentificationof pages 7-8
- veigadacunha2012molecularidentificationof pages 6-7
- veigadacunha2012molecularidentificationof pages 2-3
- key2024clppnulleukaryoteswitha pages 2-4
- veigadacunha2012molecularidentificationof pages 1-2
- veigadacunha2012molecularidentificationof pages 5-6
- donini2009recombinantproductionof pages 10-12
- https://doi.org/10.1074/jbc.M111.323485
- https://doi.org/10.3390/biom14020241
- https://platform.opentargets.org/
- https://doi.org/10.1074/jbc.m111.323485,
- https://doi.org/10.3390/biom14020241,
- https://doi.org/10.1042/bj20090748,
Lite
(PHYKPL-deep-research-lite.md)
Lite
(PHYKPL-deep-research-lite.md)PHYKPL (5-Phosphohydroxylysine Phospholyase): Enzymatic Function
PHYKPL (formerly AGXT2L2) encodes a pyridoxal-phosphate (PLP)–dependent enzyme that catalyzes the breakdown of 5-phosphohydroxylysine. Biochemical studies show that the enzyme (EC 4.2.3.134) cleaves (5R)-5-phosphonooxy-L-lysine with water to yield 2-amino-6-oxohexanoate (2-aminoadipate semialdehyde), ammonia, and inorganic phosphate[1][2]. In other words, PHYKPL’s product converts phosphorylated hydroxylysine into ammonia, phosphate and a semialdehyde (2-aminoadipate semialdehyde)[2][3]. This activity was first demonstrated by Veiga-da-Cunha et al. (2012), who produced recombinant human PHYKPL and showed it carried out exactly this reaction[3]. The enzyme is known as an ammoniophospholyase (it breaks a C–N bond releasing NH_4^+), despite its sequence similarity to aminotransferases. PHYKPL belongs to the class-III aminotransferase superfamily (the “alanine:glyoxylate aminotransferase” family) and binds PLP as cofactor[4][2]. (Sequence databases list domains IPR005814 etc., consistent with aminotransferases[5].) In line with this, UniProt/GeneCards note “pyridoxal-phosphate-dependent breakdown” of the substrate[2]. In sum, the gene product is a mitochondrial PLP-dependent lyase that catabolizes 5-phosphohydroxylysine. It localizes to the mitochondrial matrix (per prediction and antibody data[6][7]) and is synthesized from a nuclear gene transcript.
Role in Cellular and Biological Processes
PHYKPL serves at a novel juncture between collagen turnover and amino acid metabolism. Specifically, 5-hydroxylysine is an abundant collagen modification; when collagen is degraded, free 5-hydroxylysine can be phosphorylated by a dedicated kinase (HYKK, also called HYKK/AGPHD1) and then cleaved by PHYKPL[3][8]. In other words, 5-phosphohydroxylysine is a breakdown product of collagen. In mitochondria, PHYKPL (as a homotetramer) hydrolyzes this collagen-derived metabolite to feed into the lysine catabolic pathway[8]. The 2-aminoadipate semialdehyde product is a known intermediate in lysine degradation, ultimately leading to acetyl-CoA via the saccharopine/antiquitin route. Thus, PHYKPL connects extracellular matrix breakdown to energy metabolism: it helps dispose of hydroxylysine residues liberated from collagen, converting them into form that enters central metabolism. This link places PHYKPL activity in the broader context of amino-acid catabolism and collagen degradation[8]. The enzyme thereby prevents accumulation of phospho-hydroxylysine and recovers nitrogen and carbon from modified lysine residues. Consistent with this metabolic role, pathologic deficiency of PHYKPL (due to loss-of-function mutations) causes phosphohydroxylysinuria – a condition defined by elevated urinary 5-phosphohydroxylysine[6][9]. Interestingly, this biochemical phenotype appears to have no other apparent clinical manifestations[9], suggesting that the pathway is not essential for survival but is a normal part of amino acid recycling. In sum, PHYKPL’s core function is enzymatic: a PLP-dependent lyase in mitochondria that cleaves a collagen-related metabolite, linking protein (collagen) turnover to lysine/amino acid catabolism[8][3].
Gaps in Current Knowledge
-
Physiological context: It remains unclear under what conditions 5-phosphohydroxylysine is generated in vivo. While collagen degradation would release 5-hydroxylysine residues, the regulation and flux of this pathway under normal physiology or stress are poorly understood.
-
Significance of the pathway: Given that patients lacking PHYKPL activity (phosphohydroxylysinuria) show no clinical symptoms beyond metabolite buildup[9], the biological importance of this catabolic route is uncertain. It’s possible redundant disposal routes for hydroxylysine exist, or that any toxicity of accumulated metabolite is minimal.
-
Enzyme specifics: The structure, substrate specificity and kinetics of human PHYKPL have not been extensively characterized. The enzyme appears related to aminotransferases, yet its lyase activity is unusual; details of its active site and mechanism are unknown.
-
Regulation and expression: Little is known about how PHYKPL expression or activity is regulated. Tissue-specific and developmental expression patterns have not been fully mapped. (Some expression data hint at modest expression in brain and endocrine tissues, but no clear “tissue of interest” is established.)
-
Mitochondrial targeting: The mechanism of mitochondrial import of PHYKPL (e.g. targeting sequence) has not been studied.
-
Evolutionary aspects: Homologs of PHYKPL exist across vertebrates (and even in organisms like Drosophila[10]), but the evolutionary origin of this enzyme and its pathway is not well explored. It’s unclear how conserved hydroxylysine catabolism is across species.
-
Interacting partners: There is no information on whether PHYKPL forms complexes or is regulated by protein–protein interactions in mitochondria.
Questions for an Expert
-
Substrate origin: What is the main source of 5-phosphohydroxylysine in human metabolism? Is it derived solely from collagen breakdown, or might there be other pathways generating free hydroxylysine that needs disposal?
-
Redundancy: Are there alternative enzymes or pathways that can metabolize 5-phosphohydroxylysine if PHYKPL is absent? (The benign nature of phosphohydroxylysinuria hints at possible redundancy.)
-
Structure/function: What is known about the three-dimensional structure or key catalytic residues of PHYKPL? Has any structure been solved, or are there informative homology models?
-
Regulation: How is PHYKPL expression controlled? Are there known transcription factors or signaling pathways that up/downregulate this gene under specific conditions (e.g. tissue remodeling, vitamin B6 status)?
-
Tissue specificity: In which cell types or tissues is PHYKPL most active or abundant? Does it vary with development or metabolism?
-
Physiological role: Despite no overt disease, could PHYKPL have subtle roles (e.g. in nitrogen balance, redox state, or disease susceptibility)? Could there be unrecognized phenotypes in knockout models or under stress?
-
Evolution: What do comparative genomics and phylogeny tell us about PHYKPL’s origins? Are bacterial ancestors of vertebrates known to have related enzymes, and what function do they serve?
-
Potential interactions: Does PHYKPL associate with other mitochondrial enzymes (e.g. lysine catabolic enzymes) in a complex? Could it be coordinated with upstream (kinase) or downstream (dehydrogenase) steps?
Experimental Approaches to Elucidate Function
-
Biochemical assays: Express and purify human PHYKPL for in vitro enzymology. Measure its activity with synthetic 5-phosphohydroxylysine (and analogs) to determine kinetic parameters (Km, Vmax) and cofactor requirements (confirm PLP dependence)[2][1]. Test whether it can act on related substrates (e.g. different phosphoamino acids) to define specificity.
-
Cellular knockdown/knockout: Use CRISPR or siRNA in cultured human cells to disrupt PHYKPL. Then use targeted metabolomics (mass spectrometry) to detect accumulation of 5-phosphohydroxylysine or its metabolites. Compare to wild-type cells under normal and stress conditions (e.g. with added collagen fragments or excess lysine).
-
Animal models: Generate a PHYKPL-deficient mouse or zebrafish. Analyze urine and tissues for phosphohydroxylysine accumulation. Monitor for subtle phenotypes (growth, metabolism, response to high-protein diet, etc.). This could reveal in vivo roles or compensation by other pathways.
-
Metabolic tracing: Feed cells or model organisms isotopically labeled 5-hydroxylysine or collagen. Trace labeled atoms through phosphorylation by HYKK and cleavage by PHYKPL. This would confirm flux through this pathway and quantify its contribution to overall lysine catabolism.
-
Structural biology: Crystallize PHYKPL (or cryo-EM) to solve its structure. Identify active-site residues and compare to classical aminotransferases. Mutagenesis of key residues (e.g. PLP-binding lysine) could validate mechanism.
-
Expression profiling: Perform RNA/protein expression screens across tissues, development stages, or disease states. This could identify when and where PHYKPL is upregulated. Single-cell RNA-seq could pinpoint specific cell types expressing it.
-
Interaction studies: Use co-immunoprecipitation or proximity labeling (BioID) to see if PHYKPL interacts with other mitochondrial proteins or metabolic enzymes. Identifying partners might reveal regulatory mechanisms.
-
Knock-in variants: Introduce human PHYKPL mutations (e.g. those found in phosphohydroxylysinuria patients) into cell lines to study their effects on enzyme function and cell physiology.
-
Cofactor dependence: Test how cellular pyridoxal phosphate (vitamin B6) levels affect PHYKPL activity in cells, since it requires PLP[2]. Vitamin B6 deficiency might impair the enzyme and reveal latent phenotypes.
These studies together would fill gaps about PHYKPL’s substrate sources, regulation, and physiological importance. By combining biochemical, genetic, and analytical approaches, one could map the precise role of PHYKPL in amino acid and collagen metabolism and determine why its deficiency has so little clinical impact.
Sources: Information synthesized from UniProt/RefSeq summaries and enzymology references[2][6][1][3][8][9], as well as curated pathway databases.
[1] ENZYME - 4.2.3.134 5-phosphooxy-L-lysine phospho-lyase
https://enzyme.expasy.org/EC/4.2.3.134
[2] PHYKPL Gene - GeneCards | AT2L2 Protein | AT2L2 Antibody
https://www.genecards.org/cgi-bin/carddisp.pl?gene=PHYKPL
[3] [4] Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-L-lysine and phosphoethanolamine - PubMed
https://pubmed.ncbi.nlm.nih.gov/22241472/
[5] Phykpl MGI Mouse Gene Detail - MGI:1920197 - 5-phosphohydroxy-L-lysine phospholyase
https://www.informatics.jax.org/marker/MGI:1920197
[6] PHYKPL 5-phosphohydroxy-L-lysine phospho-lyase AGXT2L2 PHLU | Sigma-Aldrich
[7] PHYKPL protein expression summary - The Human Protein Atlas
https://www.proteinatlas.org/ENSG00000175309-PHYKPL
[8] [10] Reactome | PXLP-K278-PHYKPL tetramer hydrolyses 5PHL
https://reactome.org/content/detail/R-HSA-5696408
[9] Phosphohydroxylysinuria - MalaCards
https://www.malacards.org/card/phosphohydroxylysinuria?search=PHYKPL