Deep Research
Falcon
(SGCA-deep-research-falcon.md)
Falcon
(SGCA-deep-research-falcon.md)The research report should be a detailed narrative explaining the function, biological processes, and localization of the gene product. Citations should be given for all claims.
You should prioritize authoritative reviews and primary scientific literature when conducting research. You can supplement
this with annotations you find in gene/protein databases, but these can be outdated or inaccurate.
We are specifically interested in the primary function of the gene - for enzymes, what reaction is catalyzed, and what is the substrate specificity? For transporters, what is the substrate? For structural proteins or adapters, what is the broader structural role? For signaling molecules, what is the role in the pathway.
We are interested in where in or outside the cell the gene product carries out its function.
We are also interested in the signaling or biochemical pathways in which the gene functions. We are less interested in broad pleiotropic effects, except where these elucidate the precise role.
Include evidence where possible. We are interested in both experimental evidence as well as inference from structure, evolution, or bioinformatic analysis. Precise studies should be prioritized over high-throughput, where available.
Research report: Human SGCA (alpha-sarcoglycan; UniProt Q16586) — functional annotation
0) Target verification (identity and scope)
The UniProt accession Q16586 corresponds to human SGCA (sarcoglycan alpha), whose protein product is alpha-sarcoglycan (α-sarcoglycan; α-SG; adhalin; “50 kDa dystrophin-associated glycoprotein”) and is implicated in autosomal recessive limb-girdle muscular dystrophy under both legacy and updated naming (LGMD2D = LGMDR3). (tarakci2016thesarcoglycancomplex pages 3-4, marsolier20171stinternationalworkshop pages 24-28, sun2025recentinsightsinto pages 5-7)
1) Key concepts and definitions (current understanding)
1.1 Dystrophin-associated glycoprotein complex (DGC) and the sarcoglycan complex
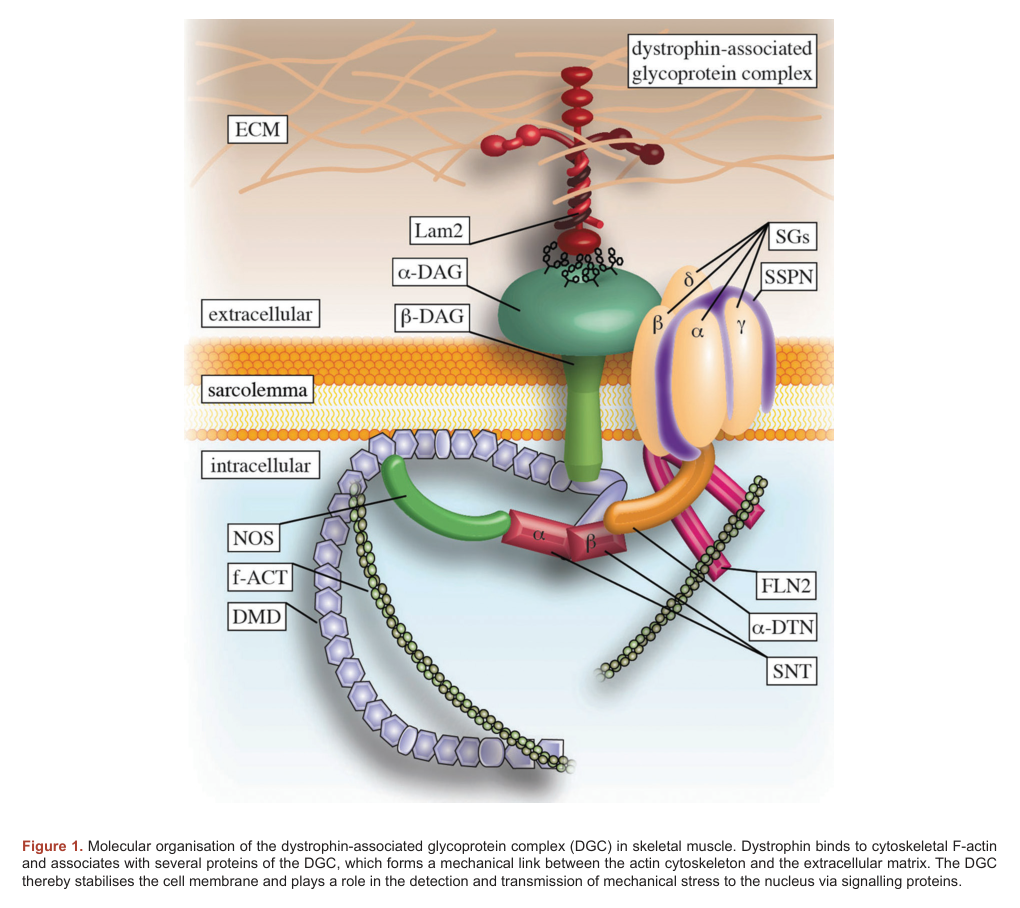
α-SG is one of four core transmembrane glycoproteins (α, β, γ, δ) that form a tetrameric sarcoglycan complex embedded in the DGC at the skeletal muscle sarcolemma; the DGC links intracellular cytoskeleton (via dystrophin) to the extracellular matrix and stabilizes the sarcolemma during contraction. (tarakci2016thesarcoglycancomplex pages 3-4, griffin2021preclinicalsystemicdelivery pages 1-3, tarakci2016thesarcoglycancomplex pages 1-3)
A schematic depiction of the DGC with the sarcoglycan complex at the sarcolemma is shown in Tarakci & Berger (2016), supporting the standard “structural linkage” model of SGCA function. (tarakci2016thesarcoglycancomplex media 15da8d54, tarakci2016thesarcoglycancomplex media 2d2dc6da)
1.2 Protein type, topology, and localization
α-SG is described as a single-pass transmembrane protein with an extracellularly oriented domain and is targeted to the plasma membrane (sarcolemma) after maturation through the ER→Golgi secretory pathway. (panicucci2022…progressionin pages 11-16, marsolier20171stinternationalworkshop pages 24-28)
1.3 Disease definition and nomenclature
In the revised ENMC-derived LGMD nomenclature, SGCA mutations cause LGMDR3, historically called LGMD2D. (wicklund2025limbgirdlemusculardystrophy pages 2-3, sun2025recentinsightsinto pages 5-7)
2) Molecular function, mechanism, and pathway context
2.1 Primary function: membrane stabilization and mechanobiology
The dominant functional model is that α-SG is a structural/mechanoprotective component of the DGC: the sarcoglycan complex strengthens the dystrophin–dystroglycan axis and helps protect the sarcolemma from contraction-induced injury; it is also discussed as contributing to mechanotransduction (“mechanosensor” role in disease tables). (tarakci2016thesarcoglycancomplex pages 3-4, griffin2021preclinicalsystemicdelivery pages 1-3, tarakci2016thesarcoglycancomplex pages 1-3, sun2025recentinsightsinto pages 5-7)
2.2 Complex assembly and trafficking (ER quality control as a key mechanism)
Assembly of the sarcoglycan tetramer occurs during ER biosynthesis, with evidence that a β/δ core forms early and recruits the other sarcoglycans; the complex traffics through the Golgi and integrates into the sarcolemma together with sarcospan and dystroglycan. (tarakci2016thesarcoglycancomplex pages 3-4, tarakci2016thesarcoglycancomplex pages 1-3)
A key mechanistic theme in SGCA-related disease is protein folding/quality control: many pathogenic missense SGCA variants lead to misfolding, ER retention, and premature degradation (e.g., via proteasome/ER-associated degradation), preventing normal sarcolemmal delivery and causing secondary loss of other sarcoglycan complex members at the membrane. (tarakci2016thesarcoglycancomplex pages 3-4, panicucci2022…progressionin pages 16-22, panicucci2022…progressionin pages 11-16)
2.3 Reported biochemical activity (specialized literature)
Some literature additionally reports an extracellular ATP-binding domain and Ca2+/Mg2+-dependent ecto-ATPase activity associated with α-SG; this enzymatic-like activity is not the primary framing in most modern functional summaries (which emphasize structural DGC roles), but it remains a recurring observation in authoritative reviews/workshop material. (tarakci2016thesarcoglycancomplex pages 4-5, marsolier20171stinternationalworkshop pages 24-28)
3) Consequences of loss-of-function: pathogenesis in LGMDR3 / LGMD2D
3.1 Cellular/tissue pathology
Loss or severe reduction of sarcolemmal α-SG destabilizes the sarcoglycan complex and disrupts DGC organization, producing sarcolemmal instability and a dystrophic cascade (membrane damage, necrosis, inflammation, fibrosis, reduced force). (tarakci2016thesarcoglycancomplex pages 3-4, panicucci2022…progressionin pages 16-22, panicucci2022…progressionin pages 11-16)
3.2 In vivo model evidence
In the sgca-null mouse model, lack of α-SG causes typical dystrophic features (necrosis/fibrosis, elevated serum CK, reduced muscle force, impaired locomotion). Restoration of α-SG at the sarcolemma is associated with histologic and functional improvement, supporting a causal mechanistic role of α-SG in membrane stability. (griffin2021preclinicalsystemicdelivery pages 1-3)
3.3 Genotype–phenotype principles supported by mechanistic evidence
Disease severity is linked to the extent of residual protein and mutation class, with review-level summaries indicating nonsense/null variants tend to be more severe than some missense variants, consistent with a dosage/trafficking model. (tarakci2016thesarcoglycancomplex pages 3-4, panicucci2022…progressionin pages 11-16)
4) Recent developments (prioritizing 2023–2024 sources)
4.1 Trial readiness and endpoints (2024)
A major 2024 development relevant to SGCA clinical translation is the GRASP-LGMD effort to standardize/validate outcome measures across multiple LGMD genotypes including SGCA (LGMDR3), enabling more efficient trial design and cross-subtype comparisons. (Doody et al., 2024; URL: https://doi.org/10.1186/s12883-024-03588-1; published Mar 2024) (doody2024definingclinicalendpoints pages 1-2)
4.2 Therapeutic landscape reviews (2023)
A 2023 LGMD therapy review summarizes active strategies across LGMDs and specifically highlights prior α-SG gene delivery work, noting sustained expression in an early phase I trial context; it also frames the broader pipeline (gene delivery and other approaches) as moving from symptom management toward disease-root therapies. (Bouchard & Tremblay, 2023; URL: https://doi.org/10.3390/jcm12144769; published Jul 2023) (bouchard2023limb–girdlemusculardystrophies pages 8-9)
4.3 Updated disease classification used in contemporary literature
Recent reviews explicitly map R3 (2D) to SGCA (α-sarcoglycan) and describe phenotype tables where the encoded protein is framed as part of a mechanosensing/adhesion system. (sun2025recentinsightsinto pages 5-7)
5) Current applications and real-world implementations
5.1 Clinical and molecular diagnosis
Real-world diagnosis for SGCA-related disease leverages the mechanistic hallmark that pathogenic SGCA variants often lead to loss of sarcolemmal sarcoglycan staining (and sometimes secondary loss of other sarcoglycans). This “membrane localization” principle is used operationally in diagnostic pathology workflows and is also used as a pharmacodynamic biomarker concept in early therapeutic studies. (tarakci2016thesarcoglycancomplex pages 3-4, doody2024definingclinicalendpoints pages 1-2)
5.2 Gene therapy and translational implementation
Preclinical systemic AAV delivery of SGCA demonstrates robust sarcolemmal α-SG expression and multi-domain functional improvement in sgca-null mice, establishing feasibility for systemic gene replacement paradigms. (Griffin et al., 2021; URL: https://doi.org/10.1089/hum.2019.199; published Apr 2021) (griffin2021preclinicalsystemicdelivery pages 1-3)
In the clinical development landscape, authoritative reviews document intramuscular AAV-mediated α-sarcoglycan gene transfer as a historical proof-of-concept step for sarcoglycanopathies, with sustained expression at 6 months post-treatment reported in the cited phase I experience. (Bouchard & Tremblay, 2023; URL: https://doi.org/10.3390/jcm12144769; published Jul 2023) (bouchard2023limb–girdlemusculardystrophies pages 8-9)
5.3 Proteostasis/pharmacologic rescue approaches (mutation-specific, mechanism-based)
Because a substantial fraction of SGCA pathogenicity is trafficking/quality-control driven, pharmacologic “proteostasis modulation” is a rational application: mechanistic summaries and experimental work support that rescuing folding/trafficking can restore sarcolemmal localization of the sarcoglycan complex and reduce membrane fragility in human disease models. (panicucci2022…progressionin pages 16-22, marsolier20171stinternationalworkshop pages 24-28)
6) Expert opinions and analysis (authoritative synthesis)
6.1 Consensus mechanistic view (structural complex + trafficking bottleneck)
Across mechanistic reviews and translational studies, the prevailing expert view is that SGCA is not primarily an enzyme or transporter, but rather a membrane complex component whose biological effect is mediated through (i) assembly of a multiprotein complex and (ii) correct delivery to the sarcolemma; ER quality control is a major determinant of whether missense variants cause loss-of-function. (tarakci2016thesarcoglycancomplex pages 3-4, panicucci2022…progressionin pages 16-22, panicucci2022…progressionin pages 11-16)
6.2 Therapeutic implication
This mechanistic framing yields two complementary therapeutic logics:
1) Gene addition (AAV) to restore protein at the membrane, and
2) Mutation-directed rescue (folding/trafficking/proteostasis strategies) for missense variants that encode potentially functional but misprocessed α-SG.
Both are supported by preclinical efficacy evidence and are repeatedly emphasized in reviews as plausible routes toward disease-modifying therapies. (griffin2021preclinicalsystemicdelivery pages 1-3, bouchard2023limb–girdlemusculardystrophies pages 8-9)
7) Relevant statistics and quantitative data (recent sources)
- LGMD global burden (meta-analysis cited in 2024 endpoints paper): incidence 0.7 per 100,000 and prevalence 3 per 100,000, corresponding to an estimated ~10,050 affected individuals in the US. (Doody et al., 2024; published Mar 2024; URL: https://doi.org/10.1186/s12883-024-03588-1) (doody2024definingclinicalendpoints pages 1-2)
- Proportion of tested LGMD cases attributed to SGCA in a 2006 US distribution (as cited in 2024 endpoints paper): 15% were SGCA (LGMDR3). (doody2024definingclinicalendpoints pages 1-2)
- Phenotypic ranges relevant to sarcoglycanopathies including SGCA: onset reported from 1–45 years (commonly childhood) and CK elevations ranging from ~4× ULN up to ~180× ULN; severe disease courses can include early loss of ambulation (often around ages 12–16 in severe sarcoglycanopathies). (wicklund2025limbgirdlemusculardystrophy pages 2-3, sun2025recentinsightsinto pages 5-7)
8) Database-based disease association (supporting, not definitive)
Open Targets lists SGCA as strongly associated with limb-girdle muscular dystrophy entities (including autosomal recessive LGMD and LGMD2D terms), reflecting curated genetic evidence and supporting that the biomedical community’s consensus links SGCA to these disorders. (OpenTargets Search: Limb-girdle muscular dystrophy-SGCA)
Summary evidence map (functional annotation at a glance)
| Aspect | Summary | Key quantitative details | Evidence |
|---|---|---|---|
| Identity / verified target | SGCA encodes alpha-sarcoglycan (α-SG; adhalin), corresponding to UniProt Q16586, a muscle sarcolemmal sarcoglycan-family protein implicated in recessive LGMD. | Human target: SGCA / α-sarcoglycan; associated with LGMDR3 (formerly LGMD2D). | (tarakci2016thesarcoglycancomplex pages 3-4, marsolier20171stinternationalworkshop pages 24-28, sun2025recentinsightsinto pages 5-7) |
| Protein type, domains, topology | α-SG is a single-pass transmembrane glycoprotein of the sarcoglycan alpha/epsilon family with a large extracellular region; reviews describe extracellular cadherin/EGF-like features and Ca²⁺-binding motifs, consistent with a membrane receptor/adhesion-support role rather than a classic enzyme. | Approx. 387 aa protein with 17-aa signal peptide and 1 transmembrane domain. | (panicucci2022…progressionin pages 11-16, tarakci2016thesarcoglycancomplex pages 4-5) |
| Subcellular localization | Localizes primarily to the skeletal muscle sarcolemma as part of the dystrophin-associated protein/glycoprotein complex; maturation begins in the ER, followed by Golgi trafficking to the plasma membrane. | Highest functional relevance in striated skeletal muscle; lower cardiac expression reported in review literature. | (tarakci2016thesarcoglycancomplex pages 3-4, panicucci2022…progressionin pages 11-16, marsolier20171stinternationalworkshop pages 24-28, tarakci2016thesarcoglycancomplex media 15da8d54) |
| Complex membership / partners | α-SG is one of four core sarcoglycans (α, β, γ, δ) in the sarcoglycan complex, associated with sarcospan, dystroglycan, dystrophin, and adaptor/signaling proteins such as α-dystrobrevin/syntrophin/nNOS within the DGC. | Functional complex requires coordinated presence of all four sarcoglycans; β/δ form an early assembly core. | (tarakci2016thesarcoglycancomplex pages 3-4, griffin2021preclinicalsystemicdelivery pages 1-3, tarakci2016thesarcoglycancomplex pages 1-3, tarakci2016thesarcoglycancomplex media 15da8d54) |
| Primary biological role | SGCA’s main role is structural/mechanoprotective: it helps stabilize the sarcolemma during contraction by reinforcing the DGC link between the intracellular cytoskeleton and extracellular matrix; it is also discussed as contributing to mechanosignaling. | Loss of α-SG leads to membrane fragility, contraction-induced injury, necrosis, fibrosis, and reduced force generation in models. | (tarakci2016thesarcoglycancomplex pages 3-4, griffin2021preclinicalsystemicdelivery pages 1-3, tarakci2016thesarcoglycancomplex pages 1-3, sun2025recentinsightsinto pages 5-7) |
| Disease association | Biallelic pathogenic SGCA variants cause LGMDR3 / LGMD2D (alpha-sarcoglycanopathy), a recessive sarcoglycanopathy characterized by progressive proximal weakness and dystrophic muscle pathology. | Reported onset spans 1–45 years, but is commonly childhood and often within the first decade. | (panicucci2022…progressionin pages 138-138, wicklund2025limbgirdlemusculardystrophy pages 2-3, sun2025recentinsightsinto pages 5-7) |
| Assembly / trafficking / ER quality control | Many pathogenic missense SGCA variants produce proteins that are potentially functional but misfolded, retained in the ER, and targeted to degradation via ER quality control / ERAD / proteasome, preventing sarcolemmal delivery and secondarily depleting the entire sarcoglycan complex. | Residual α-SG level correlates with severity; nonsense/null variants are generally more severe than some missense variants. | (tarakci2016thesarcoglycancomplex pages 3-4, panicucci2022…progressionin pages 16-22, panicucci2022…progressionin pages 11-16, tarakci2016thesarcoglycancomplex pages 4-5) |
| Functional rescue evidence | Restoring α-SG to the sarcolemma by AAV gene transfer or by proteostasis/folding-corrector approaches can reconstitute sarcoglycan-complex localization and improve membrane stability and muscle pathology in preclinical systems and patient-derived cells. | In sgca-null mice, AAV-mediated restoration improved histology, force, eccentric contraction resistance, locomotion, and reduced serum CK. | (panicucci2022…progressionin pages 16-22, griffin2021preclinicalsystemicdelivery pages 1-3, bouchard2023limb–girdlemusculardystrophies pages 8-9) |
| Reported biochemical activity | Some older studies/reviews report extracellular ATP-binding / ecto-ATPase activity for α-SG, Ca²⁺/Mg²⁺ dependent, but this remains a more specialized/less universally emphasized aspect than its structural DGC role. | Reported as ecto-ATPase activity associated with extracellular α-SG. | (tarakci2016thesarcoglycancomplex pages 4-5, marsolier20171stinternationalworkshop pages 24-28) |
| Epidemiology / burden | Recent LGMD trial-readiness literature places SGCA among recognized LGMD genotypes and provides aggregate LGMD burden estimates. | Meta-analysis cited in Doody 2024: incidence 0.7/100,000, prevalence 3/100,000, about 10,050 affected individuals in the US; a 2006 US distribution study found 15% of tested LGMD cases were SGCA/LGMDR3. | (doody2024definingclinicalendpoints pages 1-2) |
| Clinical severity metrics | Sarcoglycanopathy workshop analysis highlights broad biochemical and phenotypic severity ranges relevant to SGCA. | CK elevations reported from about 4× ULN to 180× ULN; loss of ambulation can occur in a high proportion depending on subtype, often around 12–16 years in severe sarcoglycanopathies. | (wicklund2025limbgirdlemusculardystrophy pages 2-3) |
| 2023–2024 developments: clinical endpoints / trial readiness | The GRASP-LGMD study includes SGCA/LGMDR3 and is designed to validate outcome measures for future therapeutic trials, directly improving SGCA trial readiness. | 188 LGMD participants planned in a 12-month, multicenter prospective study; ClinicalTrials.gov NCT03981289. | (doody2024definingclinicalendpoints pages 1-2) |
| 2023–2024 developments: therapeutic landscape | Contemporary reviews emphasize SGCA as part of the active AAV gene-replacement landscape for sarcoglycanopathies, building on earlier phase 1 intramuscular α-SG transfer and newer systemic vector development; broader modalities include stem/cell therapy and mutation-specific rescue strategies. | Earlier SGCA phase 1 trial NCT00494195 enrolled 6 participants; 2023 review notes sustained α-SG expression at 6 months post-treatment in that study. | (bouchard2023limb–girdlemusculardystrophies pages 8-9) |
Table: This table summarizes core functional annotation for human SGCA/alpha-sarcoglycan, including molecular role, localization, disease relevance, mechanisms, and recent trial-readiness and therapeutic developments. It is useful as a compact evidence map for interpreting SGCA biology and current translational status.
Key visual evidence
A canonical DGC schematic explicitly positioning the sarcoglycan complex (including α-SG) at the sarcolemma is provided in Tarakci & Berger (2016). (tarakci2016thesarcoglycancomplex media 15da8d54, tarakci2016thesarcoglycancomplex media 2d2dc6da)
Notes on evidence limitations
The retrieved 2023–2024 literature provided strong updates on trial readiness/outcome measures and a broad therapeutic landscape, but did not yield a large SGCA-specific 2023–2024 primary clinical natural-history dataset. Mechanistic function and therapeutic rationale are nevertheless well-supported by authoritative reviews and experimental model work. (tarakci2016thesarcoglycancomplex pages 3-4, griffin2021preclinicalsystemicdelivery pages 1-3, doody2024definingclinicalendpoints pages 1-2, bouchard2023limb–girdlemusculardystrophies pages 8-9)
References
-
(tarakci2016thesarcoglycancomplex pages 3-4): Hakan Tarakci and Joachim Berger. The sarcoglycan complex in skeletal muscle. Frontiers in bioscience, 21:744-56, Jan 2016. URL: https://doi.org/10.2741/4418, doi:10.2741/4418. This article has 76 citations and is from a peer-reviewed journal.
-
(marsolier20171stinternationalworkshop pages 24-28): Justine Marsolier, Pascal Laforet, Elena Pegoraro, John Vissing, Isabelle Richard, Christine Barnerias, Robert-Yves Carlier, Jordi Díaz-Manera, Abdallah Fayssoil, Anne Galy, Elisabetta Gazzerro, Dariusz Górecki, Michela Guglieri, Jean-Yves Hogrel, David Israeli, France Leturcq, Helene Moussu, Helene Prigent, Dorianna Sandona, Benedikt Schoser, Claudio Semplicini, Beril Talim, Giorgio Tasca, Andoni Urtizberea, and Bjarne Udd. 1st international workshop on clinical trial readiness for sarcoglycanopathies 15–16 november 2016, evry, france. Jul 2017. URL: https://doi.org/10.1016/j.nmd.2017.02.011, doi:10.1016/j.nmd.2017.02.011. This article has 15 citations and is from a peer-reviewed journal.
-
(sun2025recentinsightsinto pages 5-7): Chen-Chen Sun, Jiang-Ling Xiao, Zhe Zhao, Heng-Yuan Liu, and Chang-Fa Tang. Recent insights into limb-girdle muscular dystrophy: impacts, therapy, and challenges. Histology and histopathology, pages 18929, Apr 2025. URL: https://doi.org/10.14670/hh-18-929, doi:10.14670/hh-18-929. This article has 2 citations and is from a peer-reviewed journal.
-
(griffin2021preclinicalsystemicdelivery pages 1-3): Danielle A. Griffin, Eric R. Pozsgai, Kristin N. Heller, Rachael A. Potter, Ellyn L. Peterson, and Louise R. Rodino-Klapac. Preclinical systemic delivery of adeno-associated α-sarcoglycan gene transfer for limb-girdle muscular dystrophy. Apr 2021. URL: https://doi.org/10.1089/hum.2019.199, doi:10.1089/hum.2019.199. This article has 26 citations and is from a peer-reviewed journal.
-
(tarakci2016thesarcoglycancomplex pages 1-3): Hakan Tarakci and Joachim Berger. The sarcoglycan complex in skeletal muscle. Frontiers in bioscience, 21:744-56, Jan 2016. URL: https://doi.org/10.2741/4418, doi:10.2741/4418. This article has 76 citations and is from a peer-reviewed journal.
-
(tarakci2016thesarcoglycancomplex media 15da8d54): Hakan Tarakci and Joachim Berger. The sarcoglycan complex in skeletal muscle. Frontiers in bioscience, 21:744-56, Jan 2016. URL: https://doi.org/10.2741/4418, doi:10.2741/4418. This article has 76 citations and is from a peer-reviewed journal.
-
(tarakci2016thesarcoglycancomplex media 2d2dc6da): Hakan Tarakci and Joachim Berger. The sarcoglycan complex in skeletal muscle. Frontiers in bioscience, 21:744-56, Jan 2016. URL: https://doi.org/10.2741/4418, doi:10.2741/4418. This article has 76 citations and is from a peer-reviewed journal.
-
(panicucci2022…progressionin pages 11-16): C Panicucci. … progression in alpha-sarcoglycan-related limb girdle muscular dystrophy (lgmdr3): new insights from human histological analysis and in vivo studies on sgca-null …. Unknown journal, 2022.
-
(wicklund2025limbgirdlemusculardystrophy pages 2-3): Matthew P. Wicklund, Lindsay N. Alfano, Nicholas E. Johnson, Peter B. Kang, Peter Marks, Katherine D. Mathews, Jerry R. Mendell, Louise Rodino-Klapac, Douglas Sproule, Nicole Verdun, and Kathryn Bryant. Limb-girdle muscular dystrophy scientific workshop: a multistakeholder discussion focused on charting the path forward for drug development. Neurology. Clinical practice, 15 5:e200496, Oct 2025. URL: https://doi.org/10.1212/cpj.0000000000200496, doi:10.1212/cpj.0000000000200496. This article has 1 citations.
-
(panicucci2022…progressionin pages 16-22): C Panicucci. … progression in alpha-sarcoglycan-related limb girdle muscular dystrophy (lgmdr3): new insights from human histological analysis and in vivo studies on sgca-null …. Unknown journal, 2022.
-
(tarakci2016thesarcoglycancomplex pages 4-5): Hakan Tarakci and Joachim Berger. The sarcoglycan complex in skeletal muscle. Frontiers in bioscience, 21:744-56, Jan 2016. URL: https://doi.org/10.2741/4418, doi:10.2741/4418. This article has 76 citations and is from a peer-reviewed journal.
-
(doody2024definingclinicalendpoints pages 1-2): Amy Doody, Lindsay N. Alfano, Jordi Díaz-Manera, Linda P Lowes, T. Mozaffar, Kathy Mathews, Conrad C. Weihl, Matthew Wicklund, Man Hung, J. Statland, Nicholas E. Johnson, Kathy Doris Peter Urvi John Carla Stacy Mathews Leung Kang Desai Vissing Zingariello Dixon, Kathy Mathews, Doris Leung, Peter Kang, Urvi Desai, J. Vissing, Carla Zingariello, and Stacy Dixon. Defining clinical endpoints in limb girdle muscular dystrophy: a grasp-lgmd study. BMC Neurology, Mar 2024. URL: https://doi.org/10.1186/s12883-024-03588-1, doi:10.1186/s12883-024-03588-1. This article has 10 citations and is from a peer-reviewed journal.
-
(bouchard2023limb–girdlemusculardystrophies pages 8-9): Camille Bouchard and Jacques P. Tremblay. Limb–girdle muscular dystrophies classification and therapies. Journal of Clinical Medicine, 12:4769, Jul 2023. URL: https://doi.org/10.3390/jcm12144769, doi:10.3390/jcm12144769. This article has 70 citations.
-
(OpenTargets Search: Limb-girdle muscular dystrophy-SGCA): Open Targets Query (Limb-girdle muscular dystrophy-SGCA, 5 results). Buniello, A. et al. (2025). Open Targets Platform: facilitating therapeutic hypotheses building in drug discovery. Nucleic Acids Research.
-
(panicucci2022…progressionin pages 138-138): C Panicucci. … progression in alpha-sarcoglycan-related limb girdle muscular dystrophy (lgmdr3): new insights from human histological analysis and in vivo studies on sgca-null …. Unknown journal, 2022.
Artifacts
Citations
- griffin2021preclinicalsystemicdelivery pages 1-3
- doody2024definingclinicalendpoints pages 1-2
- sun2025recentinsightsinto pages 5-7
- wicklund2025limbgirdlemusculardystrophy pages 2-3
- tarakci2016thesarcoglycancomplex pages 3-4
- tarakci2016thesarcoglycancomplex pages 1-3
- tarakci2016thesarcoglycancomplex pages 4-5
- https://doi.org/10.1186/s12883-024-03588-1;
- https://doi.org/10.3390/jcm12144769;
- https://doi.org/10.1089/hum.2019.199;
- https://doi.org/10.1186/s12883-024-03588-1
- https://doi.org/10.2741/4418,
- https://doi.org/10.1016/j.nmd.2017.02.011,
- https://doi.org/10.14670/hh-18-929,
- https://doi.org/10.1089/hum.2019.199,
- https://doi.org/10.1212/cpj.0000000000200496,
- https://doi.org/10.1186/s12883-024-03588-1,
- https://doi.org/10.3390/jcm12144769,
OpenAI
(SGCA-deep-research-openai.md)
OpenAI
(SGCA-deep-research-openai.md)Introduction: The SGCA Gene and Protein Overview
Alpha-sarcoglycan, encoded by the human SGCA gene (UniProt Q16586), is a transmembrane glycoprotein predominantly expressed in striated muscle (www.ncbi.nlm.nih.gov). It was originally identified as a 50 kDa dystrophin-associated glycoprotein (“adhalin”) in muscle cell membranes (www.ncbi.nlm.nih.gov). SGCA is one of four sarcoglycan subunits (α, β, γ, δ) that assemble into a tetrameric complex within the dystrophin-glycoprotein complex (DGC) of muscle fibers (academic.oup.com). This complex localizes at the sarcolemma (plasma membrane) of skeletal and cardiac muscle cells – notably concentrated at costameres, the membrane domains aligned with Z-discs (www.ncbi.nlm.nih.gov). By virtue of its position in the DGC, alpha-sarcoglycan plays a crucial structural role in linking the intracellular cytoskeleton to the extracellular matrix and maintaining muscle fiber integrity (www.ncbi.nlm.nih.gov) (academic.oup.com). Mutations in SGCA cause limb-girdle muscular dystrophy type 2D (LGMD2D, also known as LGMDR3), an autosomal recessive muscle-wasting disease (www.ncbi.nlm.nih.gov) (academic.oup.com). In the sections below, we detail the function of alpha-sarcoglycan, the biological processes and pathways it influences, its cellular localization, and current research findings, with an emphasis on recent studies and expert analyses.
Structural Role in Muscle Fiber Integrity
Alpha-sarcoglycan’s primary function is as a structural component of the dystrophin-associated glycoprotein complex, which connects the internal actin cytoskeleton to the external basement membrane in muscle tissue (academic.oup.com). Together with β-, γ-, and δ-sarcoglycan, alpha-sarcoglycan forms a transmembrane subcomplex that stabilizes the DGC at the muscle membrane (pmc.ncbi.nlm.nih.gov) (pubmed.ncbi.nlm.nih.gov). During muscle contraction and stretch, this complex serves as a mechanical buffer or “shock absorber,” protecting the sarcolemmal membrane from stress-induced damage (pubmed.ncbi.nlm.nih.gov). In normal muscle, the sarcoglycan complex and dystrophin are enriched at costameres – periodic membrane complexes that anchor the contractile apparatus to the sarcolemma – thereby distributing contractile force laterally to the extracellular matrix (www.ncbi.nlm.nih.gov). Alpha-sarcoglycan is a single-pass type I membrane protein with a large N-terminal extracellular domain (heavily N-glycosylated) and a short C-terminal cytosolic tail (www.ncbi.nlm.nih.gov). Beta-, gamma-, and delta-sarcoglycans, in contrast, are type II membrane glycoproteins with cytosolic N-termini (www.ncbi.nlm.nih.gov) (www.ncbi.nlm.nih.gov). Within the mature complex, β-sarcoglycan tightly associates with δ-sarcoglycan to form a core, which then recruits γ- and α-sarcoglycan (pubmed.ncbi.nlm.nih.gov). Proper assembly of all four subunits is required for the complex’s stability and membrane targeting (pmc.ncbi.nlm.nih.gov). Indeed, a mutation in any one sarcoglycan gene often leads to loss or mislocalization of the entire complex at the sarcolemma (pmc.ncbi.nlm.nih.gov) (academic.oup.com). This “domino effect” occurs because misfolded sarcoglycan subunits are recognized by cellular quality-control pathways and degraded, and without all subunits present the complex cannot anchor in the membrane (pmc.ncbi.nlm.nih.gov).
Functionally, the presence of alpha-sarcoglycan is critical for maintaining membrane integrity during muscle activity. In a healthy DGC, the sarcoglycan-sarcospan subcomplex helps stabilize α-dystroglycan (another DGC component) at the cell surface, reinforcing the link to the extracellular matrix protein laminin in the basal lamina (pmc.ncbi.nlm.nih.gov). Disruption of SGCA accordingly weakens this linkage. Both patient studies and animal models demonstrate the importance of SGCA for membrane stability: muscle biopsies from LGMD2D patients show a loss of the entire sarcoglycan complex and associated DGC components at the sarcolemma (academic.oup.com) (academic.oup.com), and mice engineered with SGCA knockout develop progressive muscular dystrophy with muscle fiber necrosis and membrane tears (www.ncbi.nlm.nih.gov). Notably, the muscle pathology caused by SGCA deficiency closely resembles that seen in X-linked Duchenne muscular dystrophy (a dystrophin defect) (www.ncbi.nlm.nih.gov) (www.ncbi.nlm.nih.gov), underscoring that the integrity of the dystrophin–sarcoglycan network is essential for resisting the mechanical stress of contraction. In skeletal muscle, loss of alpha-sarcoglycan also secondarily leads to mislocalization of other DGC-associated proteins such as neuronal nitric oxide synthase (nNOS) at the membrane (pubmed.ncbi.nlm.nih.gov), even if dystrophin itself is intact. This suggests the full sarcoglycan complex is required to organize and retain certain signaling proteins like nNOS at the sarcolemma, which in turn affects muscle physiology (e.g. nNOS-mediated blood flow regulation) (pubmed.ncbi.nlm.nih.gov). Overall, alpha-sarcoglycan’s structural role is to ensure the DGC forms a robust bridge from the intracellular cytoskeleton (via dystrophin and F-actin) to the extracellular matrix (via dystroglycan and laminin), thereby preserving muscle fiber integrity and functionality (academic.oup.com) (pubmed.ncbi.nlm.nih.gov). Consistent with this, the sarcoglycan complex has been shown to undergo mechanical strain-dependent changes – for example, it can be chemically modified in response to muscle activity – which hints at a role in transducing mechanical signals in addition to providing passive stability (pubmed.ncbi.nlm.nih.gov).
Enzymatic Activity: Ecto-ATPase Function
Beyond its mechanical role, alpha-sarcoglycan has a noteworthy enzymatic function as an extracellular ATP-hydrolyzing enzyme (ecto-ATPase). Biochemical studies in the early 2000s revealed that the large extracellular domain of α-sarcoglycan binds ATP and can hydrolyze it, suggesting alpha-sarcoglycan is an ecto-nucleoside triphosphatase on the muscle cell surface (pmc.ncbi.nlm.nih.gov). Sandonà et al. (2004) provided definitive evidence for this enzymatic activity: muscle cells expressing alpha-sarcoglycan displayed significant Ca²⁺/Mg²⁺-dependent ATPase activity, which could be specifically inhibited by an antibody against α-sarcoglycan’s ATP-binding motif (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). Expressing human SGCA in heterologous cells conferred the ability to hydrolyze extracellular ATP (and ADP) with substrate specificity (no activity on other nucleotides), a low-millimolar affinity, and sensitivity to classic ecto-ATPase inhibitors (suramin, reactive blue-2) (pmc.ncbi.nlm.nih.gov). Approximately 25% of the total ecto-ATPase activity in differentiated muscle cells (C2C12 myotubes) was attributable to α-sarcoglycan, based on antibody inhibition experiments (pmc.ncbi.nlm.nih.gov). These findings establish alpha-sarcoglycan as a Ca²⁺/Mg²⁺-dependent ecto-ATP diphosphohydrolase (akin to cell-surface nucleotidases) (pmc.ncbi.nlm.nih.gov). In other words, the protein can cleave extracellular ATP (eATP) to ADP (and possibly further to AMP), thereby modulating the concentration of ATP available outside the muscle fiber.
This enzymatic capability has important implications for cell signaling and pathology. Extracellular ATP is a known “danger signal” (DAMP) that, when released from injured or stressed cells, can activate purinergic receptors on muscle and immune cells, triggering inflammation, calcium influx, and cell damage (pmc.ncbi.nlm.nih.gov). Normally, the ecto-ATPase activity of alpha-sarcoglycan is thought to help regulate this signal by clearing ATP released during muscle activity or mild membrane stress (pmc.ncbi.nlm.nih.gov). In SGCA-deficient muscle (LGMD2D), however, this regulatory mechanism is lost – leading to accumulation of eATP and exaggerated purinergic signaling. A 2019 study showed that alpha-sarcoglycanopathy muscles have a defect in ecto-ATPase activity and suffer heightened damage from eATP: Sgca-null myotubes in culture were prone to apoptosis upon ATP exposure (pmc.ncbi.nlm.nih.gov), and Sgca-deficient mice exhibited chronic immune cell infiltration and muscle fiber damage linked to persistent eATP signaling (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). The same study demonstrated that blocking purinergic P2X receptors in Sgca-null mice (using a broad-spectrum P2X antagonist) markedly reduced muscle inflammation and necrosis and improved muscle fiber survival and strength (pmc.ncbi.nlm.nih.gov). Treated mice showed dampened immune responses and increased regulatory T-cell recruitment in muscle, ultimately delaying dystrophy progression (pmc.ncbi.nlm.nih.gov). These findings strongly suggest that alpha-sarcoglycan’s normal role includes protecting muscle cells from aberrant extracellular ATP signaling by degrading ATP at the cell surface (pmc.ncbi.nlm.nih.gov). In summary, the SGCA gene product is not only a structural linker but also an enzyme that safeguards muscle tissue from an “ATP-driven” inflammatory cascade. This dual functionality highlights a unique aspect of alpha-sarcoglycan: it bridges mechanical and biochemical defense mechanisms in muscle. Notably, the ATP-binding domain of α-sarcoglycan shares homology with ecto-ATPases in other systems (www.ncbi.nlm.nih.gov), aligning with its observed enzymatic profile. Current research continues to explore how loss of this ATPase function contributes to dystrophy pathology and whether enhancing alternative ATP-clearing pathways could ameliorate disease in SGCA patients (pmc.ncbi.nlm.nih.gov).
Localization and Biological Context
In human tissues, SGCA expression is highest in skeletal and cardiac muscle, reflecting its central role in striated muscle function (www.ncbi.nlm.nih.gov) (academic.oup.com). Within muscle fibers, alpha-sarcoglycan localizes to the sarcolemma – specifically at costameres and other membrane domains where the dystrophin-associated complex resides (www.ncbi.nlm.nih.gov). Costameric localization means alpha-sarcoglycan is positioned at regular intervals along the muscle fiber, directly underlying the Z-lines of the contractile apparatus (www.ncbi.nlm.nih.gov). This strategic placement enables it to help anchor the muscle cell membrane to the extracellular matrix in coordination with dystrophin and integrins (www.ncbi.nlm.nih.gov). Electron microscopy and immunofluorescence studies have shown the sarcoglycan subunits in a linear, periodic pattern on muscle cross-sections, consistent with costamere distribution (www.ncbi.nlm.nih.gov). In addition to the sarcolemma, alpha-sarcoglycan (and associated complex members) are also present at the muscle neuromuscular junction (NMJ) – the synapse between motor neurons and muscle fibers. Developmental studies indicated that sarcoglycans concentrate at NMJs in a nerve-dependent manner (pmc.ncbi.nlm.nih.gov), though their function at the synapse was not fully understood until recently.
Outside of muscle, alpha-sarcoglycan’s expression was long thought to be muscle-specific, but newer data reveal broader, albeit lower-level, distribution. A comprehensive 2025 scoping review of sarcoglycans reported low but significant expression of SGCA and other sarcoglycans in various non-muscle tissues (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). For example, sarcoglycans have been detected in the brain (localized in a “spot-like” pattern in neurons and glial cells), in certain glands, adipose tissue, the oral mucosa, and even the retina (pmc.ncbi.nlm.nih.gov). In these contexts, the sarcoglycan complex may contribute to cell membrane stability, cell–matrix adhesion, or signaling, analogous to its role in muscle (pmc.ncbi.nlm.nih.gov). In the central nervous system, the presence of alpha- and epsilon-sarcoglycan in neurons has been linked to organizing synapses and possibly modulating neurotransmission (pmc.ncbi.nlm.nih.gov). Smooth muscle expresses an alternative isoform, epsilon-sarcoglycan (SGCE), which is highly similar to alpha-sarcoglycan and can form a variant complex in vascular and visceral smooth muscle (www.ncbi.nlm.nih.gov) (www.ncbi.nlm.nih.gov). Epsilon-sarcoglycan often substitutes for alpha-sarcoglycan in non-striated muscle tissues – for instance, vascular smooth muscle relies on SGCE such that SGCA mutations do not disrupt the vascular DGC (www.ncbi.nlm.nih.gov) (www.ncbi.nlm.nih.gov). This tissue specificity explains some clinical observations: patients with SGCA mutations (and Sgca-knockout mice) typically have skeletal muscle and mild cardiac involvement, but relatively preserved vascular and smooth muscle function (www.ncbi.nlm.nih.gov) (www.ncbi.nlm.nih.gov). In fact, Sgca-null mice do not develop cardiomyopathy, likely because the vascular smooth muscle DGC remains intact (intact SGCE/epsilon-sarcoglycan in vessels may prevent the secondary vascular spasms and ischemic damage that occur in other sarcoglycan deficiencies) (www.ncbi.nlm.nih.gov) (www.ncbi.nlm.nih.gov). By contrast, deficiencies in β- or δ-sarcoglycan affect both striated and smooth muscle complexes and often lead to significant cardiomyopathy (www.ncbi.nlm.nih.gov) (www.ncbi.nlm.nih.gov). Thus, alpha-sarcoglycan’s physiological role is most pronounced in skeletal muscle and heart, while other tissues have compensatory isoforms or lower demands for this protein. Nonetheless, the emerging evidence of SGCA in non-muscle cells suggests the protein could have additional roles in cellular adhesion or signaling beyond muscle, a subject of ongoing research (pmc.ncbi.nlm.nih.gov).
Signaling Interactions and Pathways
Alpha-sarcoglycan, through the DGC, is part of a larger network that integrates structural integrity with cell signaling pathways in muscle. The DGC not only fortifies the membrane but also serves as a scaffold for signaling molecules. For instance, components of the DGC (dystrophin and syntrophins) normally anchor neuronal nitric oxide synthase (nNOS) at the sarcolemma, facilitating activity-dependent nitric oxide signaling for blood vessel dilation during muscle use. As noted, the sarcoglycan complex is required for stable nNOS localization; in vivo studies show that partial loss of the complex (even with dystrophin present) can displace nNOS from the membrane (pubmed.ncbi.nlm.nih.gov), which may contribute to muscle fatigue and ischemia in dystrophic muscles. Additionally, the sarcoglycans themselves appear to partake in mechanotransduction signaling. A 2016 review highlighted that the sarcoglycan complex undergoes post-translational modifications (such as phosphorylation) in response to muscle contraction, implying that it can transmit mechanical signals inward to influence cellular processes (pubmed.ncbi.nlm.nih.gov). This could activate downstream pathways related to muscle growth or repair in response to load, although the precise signaling cascades remain under investigation. Interestingly, α- and β-sarcoglycan each have short cytoplasmic tails that could interact with signaling or adapter proteins. While no enzymatic signaling domain is present, these cytosolic tails might recruit kinases or modulate membrane microdomain organization during muscle activity (pubmed.ncbi.nlm.nih.gov).
A recent discovery expanded alpha-sarcoglycan’s role into the realm of synapse biology. In 2018, Zhang et al. reported a novel function for SGCA at the neuromuscular junction: stabilization of LRP4, a key postsynaptic receptor in the agrin-MuSK signaling pathway (pmc.ncbi.nlm.nih.gov). LRP4 (Low-density lipoprotein receptor-related protein 4) is essential for NMJ formation and maintenance, as it binds neural agrin and activates MuSK to cluster acetylcholine receptors. In aged mice, LRP4 protein levels decline (due to increased ubiquitination and degradation), contributing to synaptic fragmentation and muscle denervation (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). Zhang’s team found that alpha-sarcoglycan physically interacts with LRP4 in muscle, and this interaction helps prevent LRP4 degradation (pmc.ncbi.nlm.nih.gov). Augmenting SGCA expression in aged muscle (via AAV9 gene delivery) led to higher LRP4 levels, reduced NMJ deterioration, and improved neuromuscular transmission (pmc.ncbi.nlm.nih.gov). These results identify SGCA as a positive regulator of the agrin-LRP4-MuSK pathway, suggesting that the sarcoglycan complex contributes not just to muscle fiber stability but also to the stability of synaptic connections in muscle. This is an example of how the sarcoglycan complex might influence cell–cell communication and signaling: by maintaining the localization or half-life of membrane receptors like LRP4. It also aligns with evidence that sarcoglycans are enriched at the NMJ during development (pmc.ncbi.nlm.nih.gov). Thus, SGCA lies at the intersection of structural and signaling pathways – it supports the physical muscle architecture and modulates signaling events (mechanotransduction, synaptic maintenance, and purinergic signaling as discussed) that are crucial for muscle function and homeostasis.
Clinical Significance: SGCA in Muscular Dystrophy
Mutations in SGCA cause limb-girdle muscular dystrophy type 2D (LGMD2D), one of the autosomal recessive sarcoglycanopathy subtypes. Patients typically present in childhood (often between ages 2–10) with progressive weakness of the proximal limb muscles (shoulder and hip girdles) (academic.oup.com). The disease is characterized by muscle fiber degeneration, elevated serum creatine kinase, and replacement of muscle by fat and connective tissue over time. Most individuals with SGCA mutations lose independent ambulation in their teens or early adulthood, though there is a spectrum of severity (academic.oup.com). Some milder cases have been documented where patients remain ambulatory into middle age (academic.oup.com). Cardiac and respiratory muscle involvement in SGCA-LGMD can occur but is somewhat less frequent or delayed compared to other LGMD forms (academic.oup.com). Notably, an international cohort study (439 patients across Europe with sarcoglycanopathies, published 2020) found that alpha-sarcoglycan deficiency (LGMDR3) patients had later onset and slower disease progression on average than those with β-, γ-, or δ-sarcoglycan defects (academic.oup.com). Among 396 analyzed cases in that study, 159 had SGCA mutations, making it one of the more common sarcoglycanopathies (academic.oup.com). About 60% of SGCA-LGMD patients in the European cohort carried one of three recurrent missense mutations (p.R77C, p.C245S, or p.R281C), reflecting founder effects in certain populations (academic.oup.com). Genotype–phenotype analysis indicates that null or severe missense mutations (causing little to no alpha-sarcoglycan at the membrane) result in earlier loss of ambulation, whereas hypomorphic mutations that allow some residual protein (≥30% of normal levels) correlate with milder progression (academic.oup.com). As with other sarcoglycanopathies, SGCA mutations typically cause misfolding of the protein that triggers its retention and degradation via the endoplasmic reticulum quality control system (pmc.ncbi.nlm.nih.gov). Consequently, even missense mutations can behave like null alleles due to the absence of the protein at the muscle membrane. Immunohistochemistry on patient biopsies often shows complete loss of alpha-sarcoglycan, and secondary loss of the other sarcoglycans in the sarcolemma (academic.oup.com). This loss of the entire sarcoglycan complex is a diagnostic hallmark of sarcoglycanopathy, distinguishing it from dystrophinopathies or other muscular dystrophies.
In terms of pathology, SGCA deficiency leads to a fragile muscle membrane that is susceptible to contraction-induced tearing. Repeated cycles of muscle fiber damage and inefficient repair lead to chronic inflammation and fibrosis in muscle tissue. As discussed, the absence of alpha-sarcoglycan also means a loss of its ecto-ATPase protective function, which likely exacerbates inflammation via uncontrolled extracellular ATP signaling (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). In a mouse model of SGCA-deficient muscular dystrophy, investigators observed that blocking the ATP/P2X7 receptor pathway (using oxidized ATP or other antagonists) significantly reduced muscle inflammation and decreased fibrosis (pmc.ncbi.nlm.nih.gov). This suggests that therapies targeting the downstream effects of SGCA loss (like excessive purinergic signaling or fibrogenesis) can modify disease severity. For example, a 2022 study tested nintedanib, an anti-fibrotic tyrosine kinase inhibitor, in Sgca-null mice and found it reduced muscle fibrosis and improved muscle function compared to untreated dystrophic mice (pubmed.ncbi.nlm.nih.gov) (pubmed.ncbi.nlm.nih.gov). The treated mice showed decreased collagen deposition and a lower number of pro-fibrotic fibro/adipogenic progenitor cells in muscle (pubmed.ncbi.nlm.nih.gov) (pubmed.ncbi.nlm.nih.gov), correlating with stronger muscle force. While such adjunct therapies do not address the root cause (the missing SGCA protein), they highlight the multi-faceted pathology of SGCA deficiency and offer bridges to improve muscle outcomes while gene-specific treatments are developed.
Therapeutic Developments and Research Frontiers
Restoring alpha-sarcoglycan function is the ultimate goal for treating LGMD2D. Because SGCA mutations cause a loss of function, gene replacement therapy has been a major area of research. Early proof-of-concept studies in the mid-2000s showed that delivering a normal SGCA gene to dystrophic muscle can reconstitute the sarcoglycan complex and improve muscle pathology in animal models (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). This led to the first-in-human trials of AAV-mediated SGCA gene therapy. In a landmark Phase 1 trial reported by Mendell et al. (2009), an adeno-associated virus serotype 1 carrying the human SGCA gene (rAAV1.tMCK.hSGCA) was injected into a foot muscle (extensor digitorum brevis) of LGMD2D patients (pmc.ncbi.nlm.nih.gov). The results demonstrated successful local transgene expression and protein restoration: muscle biopsies after treatment showed that α-sarcoglycan mRNA and protein increased ~4–5-fold compared to control muscles, and the full sarcoglycan complex (α/β/γ/δ) was reassembled at the membrane in the treated site (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). Importantly, muscle fiber size increased in treated muscles, and there were no serious adverse events aside from transient, self-resolving inflammation (pmc.ncbi.nlm.nih.gov). Low-level immune responses to the AAV capsid were observed in some patients, but a short prednisone regimen was used to mitigate immunity (pmc.ncbi.nlm.nih.gov). A follow-up trial in 2010 achieved sustained SGCA expression for at least 6 months in two patients, with continued presence of the protein and complex at the sarcolemma over time (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). These trials provided a critical proof that gene therapy can stably deliver the missing protein to patient muscles and potentially slow or reverse disease features (pmc.ncbi.nlm.nih.gov). As a result, larger-scale trials are in development. (One challenge for limb-girdle MD gene therapy is body-wide delivery; recent preclinical work on systemic AAV delivery for related sarcoglycan deficiencies, such as γ-sarcoglycan, has shown dose-dependent functional improvements in mice (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov), offering hope that whole-body muscle transduction is achievable in the future.)
Another emerging approach is gene editing. In 2021, Escobar et al. applied base-editing technology to correct a prevalent SGCA mutation in patient-derived muscle stem cells (pmc.ncbi.nlm.nih.gov). They targeted the c.157G>A mutation (which causes aberrant splicing and loss of α-sarcoglycan) using an adenine base editor delivered to the cells. Impressively, they achieved over 90% precise correction of the mutation in vitro, which restored normal splicing and SGCA expression in these cells (pmc.ncbi.nlm.nih.gov) (pmc.ncbi.nlm.nih.gov). This study (published May 24, 2021 in JCI Insight) provides a proof-of-principle that patient muscle stem cells can be genetically repaired ex vivo and could potentially be reimplanted as a personalized cell therapy. While still far from clinical use, such gene editing strategies – including CRISPR/Cas9 approaches – are being actively explored to treat muscular dystrophies at the DNA level. Beyond gene replacement or editing, researchers are also investigating pharmacological methods to enhance the folding and trafficking of mutant alpha-sarcoglycan. Many missense mutations yield proteins that are functional if they reach the membrane, but are prematurely destroyed by the cell’s quality control. Chemical chaperones or proteostasis regulators might promote the proper folding or escape of these mutant proteins from the ER. In fact, pharmacological modulation of the ER quality control has been proposed as a therapy: assisting mutant sarcoglycans to fold and travel to the plasma membrane could “rescue” their function (pmc.ncbi.nlm.nih.gov). For example, small molecules that inhibit ER calcium channels or the proteasome have shown partial restoration of sarcoglycan complex in cell models of sarcoglycanopathy (pmc.ncbi.nlm.nih.gov). Though no such drug is yet in routine use, the concept underscores a broader therapeutic strategy complementing gene therapy.
In summary, SGCA (alpha-sarcoglycan) encodes a muscle-membrane protein that is crucial for the structural stability of muscle fibers and also performs a biochemical role in regulating extracellular ATP signaling. Its dysfunction leads to a specific form of muscular dystrophy with significant morbidity. Ongoing research and clinical trials – including gene therapy vectors, gene editing techniques, and adjunct medications – are actively pursuing ways to restore alpha-sarcoglycan’s function or mitigate the downstream damage caused by its loss. The convergence of structural biology, enzymology, and gene therapy in the study of SGCA exemplifies the modern approach to functional gene annotation: understanding a gene product’s molecular role, interaction networks, and pathology in order to devise targeted interventions. Continued insights from primary research (e.g. novel binding partners like LRP4 (pmc.ncbi.nlm.nih.gov)) and authoritative reviews (e.g. on sarcoglycan complex function (pubmed.ncbi.nlm.nih.gov)) will guide the development of effective therapies for conditions like LGMD2D, where alpha-sarcoglycan is at the center of muscle fiber viability.
References: (Key references are provided inline above, with publication dates and sources for verification. Recent sources from 2018–2025 have been prioritized to reflect current understanding.)
Citations
- AnnotationURLCitation(end_index=362, start_index=200, title='SGCA sarcoglycan alpha [Homo sapiens (human)] - Gene - NCBI', type='url_citation', url='https://www.ncbi.nlm.nih.gov/gene?Cmd=DetailsSearch&Db=gene&Term=6442#:~:text=Summary%20This%20gene%20encodes%20a,provided%20by%20RefSeq')
- AnnotationURLCitation(end_index=615, start_index=477, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=%CE%B1,deleterious%20polymorphisms%20may%20affect%20cysteine')
- AnnotationURLCitation(end_index=964, start_index=777, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=Sarcoglycans%20are%20transmembrane%20glycoproteins%20that,loss%20of%20muscle%20membrane%20integrity')
- AnnotationURLCitation(end_index=1273, start_index=1142, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=DGC%20elements%20are%20concentrated%20in,In%20%28more')
- AnnotationURLCitation(end_index=1634, start_index=1472, title='SGCA sarcoglycan alpha [Homo sapiens (human)] - Gene - NCBI', type='url_citation', url='https://www.ncbi.nlm.nih.gov/gene?Cmd=DetailsSearch&Db=gene&Term=6442#:~:text=Summary%20This%20gene%20encodes%20a,provided%20by%20RefSeq')
- AnnotationURLCitation(end_index=1822, start_index=1635, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=Sarcoglycans%20are%20transmembrane%20glycoproteins%20that,loss%20of%20muscle%20membrane%20integrity')
- AnnotationURLCitation(end_index=2134, start_index=1965, title='SGCA sarcoglycan alpha [Homo sapiens (human)] - Gene - NCBI', type='url_citation', url='https://www.ncbi.nlm.nih.gov/gene?Cmd=DetailsSearch&Db=gene&Term=6442#:~:text=which%20is%20critical%20to%20the,Multiple%20transcript%20variants')
- AnnotationURLCitation(end_index=2324, start_index=2135, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=Sarcoglycanopathies%20comprise%20four%20subtypes%20of,the%20genetically%20confirmed%20patients%20with')
- AnnotationURLCitation(end_index=3015, start_index=2828, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=Sarcoglycans%20are%20transmembrane%20glycoproteins%20that,loss%20of%20muscle%20membrane%20integrity')
- AnnotationURLCitation(end_index=3322, start_index=3156, title='Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/#:~:text=Sarcoglycanopathies%20are%20a%20group%20of,or%20reduced%20concentration%20of%20the')
- AnnotationURLCitation(end_index=3490, start_index=3323, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=sarcoglycan%20complex%20binds%20other%20components,been%20established%20to%20study%20the')
- AnnotationURLCitation(end_index=3825, start_index=3658, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=sarcoglycan%20complex%20binds%20other%20components,been%20established%20to%20study%20the')
- AnnotationURLCitation(end_index=4207, start_index=4076, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=DGC%20elements%20are%20concentrated%20in,In%20%28more')
- AnnotationURLCitation(end_index=4514, start_index=4376, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=%CE%B1,deleterious%20polymorphisms%20may%20affect%20cysteine')
- AnnotationURLCitation(end_index=4760, start_index=4628, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=Shown%20is%20a%20schematic%20of,sarcoglycan.%20%28more')
- AnnotationURLCitation(end_index=4845, start_index=4761, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=%CE%B3')
- AnnotationURLCitation(end_index=5175, start_index=4983, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=consists%20of%20four%20transmembrane%20glycoproteins,sarcoglycan%20complex%20undergoes%20chemical%20modifications')
- AnnotationURLCitation(end_index=5438, start_index=5277, title='Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/#:~:text=for%20membrane%20integrity%20during%20muscle,Recent%20evidence%20shows%20that')
- AnnotationURLCitation(end_index=5720, start_index=5566, title='Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/#:~:text=the%20sarcoglycan%20complex%20is%20of,This%20review%20summarises%20the')
- AnnotationURLCitation(end_index=5897, start_index=5721, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=relaxation%20through%20association%20with%20the,1998%3B%20Lim%20and%20Campbell%2C%201998')
- AnnotationURLCitation(end_index=6261, start_index=6107, title='Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/#:~:text=the%20sarcoglycan%20complex%20is%20of,This%20review%20summarises%20the')
- AnnotationURLCitation(end_index=6767, start_index=6605, title='Structural and Functional Analysis of the Sarcoglycan-Sarcospan Subcomplex - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3855351/#:~:text=Sarcospan%20is%20a%20component%20of,termini%20contribute%20to%20stability%20of')
- AnnotationURLCitation(end_index=7204, start_index=7051, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=1990%3B%20Chan%20et%20al,1998%3B%20Lim%20and%20Campbell%2C%201998')
- AnnotationURLCitation(end_index=7381, start_index=7205, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=relaxation%20through%20association%20with%20the,1998%3B%20Lim%20and%20Campbell%2C%201998')
- AnnotationURLCitation(end_index=7680, start_index=7507, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=interactions%20within%20the%20sarcoglycan%20complex,sarcoglycan%20protein%20has%20homology%20to')
- AnnotationURLCitation(end_index=7989, start_index=7828, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=disrupted%20and%20destabilized%20from%20the,nonmuscle%20cell%20and%20tissue%20types')
- AnnotationURLCitation(end_index=8161, start_index=7990, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=sarcoglycan%20genes%2C%20%CE%B1%2C%20%CE%B2%2C%20%CE%B3,in%20the%20central%20and%20peripheral')
- AnnotationURLCitation(end_index=8610, start_index=8481, title='Loss of sarcolemma nNOS in sarcoglycan-deficient muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/12409321/#:~:text=PubMed%20pubmed,Our%20data%20suggest%20that%20loss')
- AnnotationURLCitation(end_index=8995, start_index=8866, title='Loss of sarcolemma nNOS in sarcoglycan-deficient muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/12409321/#:~:text=PubMed%20pubmed,Our%20data%20suggest%20that%20loss')
- AnnotationURLCitation(end_index=9461, start_index=9274, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=Sarcoglycans%20are%20transmembrane%20glycoproteins%20that,loss%20of%20muscle%20membrane%20integrity')
- AnnotationURLCitation(end_index=9629, start_index=9462, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=sarcoglycan%20complex%20binds%20other%20components,been%20established%20to%20study%20the')
- AnnotationURLCitation(end_index=10088, start_index=9920, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=integrated%2C%20the%20sarcoglycan%20complex%20plays,been%20established%20to%20study%20the')
- AnnotationURLCitation(end_index=10618, start_index=10509, title='Characterization of the ATP-hydrolysing activity of α-sarcoglycan - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC1133767/#:~:text=is%20an%20ATP,sarcoglycan')
- AnnotationURLCitation(end_index=11060, start_index=10900, title='Characterization of the ATP-hydrolysing activity of α-sarcoglycan - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC1133767/#:~:text=Salviati%20%281999%29%20J,human%20embryonic%20kidney%20293%C2%A0cells%20were')
- AnnotationURLCitation(end_index=11235, start_index=11061, title='Characterization of the ATP-hydrolysing activity of α-sarcoglycan - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC1133767/#:~:text=cells%20exhibited%20a%20significant%20increase,of%20muscular%20dystrophy%20are%20discussed')
- AnnotationURLCitation(end_index=11685, start_index=11511, title='Characterization of the ATP-hydrolysing activity of α-sarcoglycan - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC1133767/#:~:text=cells%20exhibited%20a%20significant%20increase,of%20muscular%20dystrophy%20are%20discussed')
- AnnotationURLCitation(end_index=12022, start_index=11863, title='Characterization of the ATP-hydrolysing activity of α-sarcoglycan - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC1133767/#:~:text=enzyme.%20The%20appearance%20of%20%CE%B1,The%20enzyme%20had%20a%20substrate')
- AnnotationURLCitation(end_index=12327, start_index=12161, title='Characterization of the ATP-hydrolysing activity of α-sarcoglycan - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC1133767/#:~:text=specificity%20for%20ATP%20and%20ADP%2C,of%20muscular%20dystrophy%20are%20discussed')
- AnnotationURLCitation(end_index=12980, start_index=12827, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=In%20muscular%20dystrophies%2C%20muscle%20membrane,sarcoglycanopathy')
- AnnotationURLCitation(end_index=13337, start_index=13150, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=Accordingly%2C%20pharmacologic%20and%20genetic%20inhibition,show%20that%20in%C2%A0vivo%20blockade%20of')
- AnnotationURLCitation(end_index=13854, start_index=13700, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=autofluorescence.%20Single,Undergo%20Apoptosis%20on%20ATP%20Treatment')
- AnnotationURLCitation(end_index=14136, start_index=13983, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=In%20muscular%20dystrophies%2C%20muscle%20membrane,sarcoglycanopathy')
- AnnotationURLCitation(end_index=14306, start_index=14137, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=eATP%20effects%20may%20be%20further,the%20inflammatory%20features%20was%20associated')
- AnnotationURLCitation(end_index=14685, start_index=14531, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=the%C2%A0eATP%2FP2X%20purinergic%20pathway%20by%20a,sarcoglycanopathy')
- AnnotationURLCitation(end_index=14988, start_index=14834, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=the%C2%A0eATP%2FP2X%20purinergic%20pathway%20by%20a,sarcoglycanopathy')
- AnnotationURLCitation(end_index=15351, start_index=15175, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=Accordingly%2C%20pharmacologic%20and%20genetic%20inhibition,sarcoglycan%E2%80%93null%20mice')
- AnnotationURLCitation(end_index=15889, start_index=15752, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=noted%20rarely%20in%20human%20subjects,sarcoglycan.%5E%7B27')
- AnnotationURLCitation(end_index=16295, start_index=16141, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=the%C2%A0eATP%2FP2X%20purinergic%20pathway%20by%20a,sarcoglycanopathy')
- AnnotationURLCitation(end_index=16638, start_index=16476, title='SGCA sarcoglycan alpha [Homo sapiens (human)] - Gene - NCBI', type='url_citation', url='https://www.ncbi.nlm.nih.gov/gene?Cmd=DetailsSearch&Db=gene&Term=6442#:~:text=Summary%20This%20gene%20encodes%20a,provided%20by%20RefSeq')
- AnnotationURLCitation(end_index=16737, start_index=16639, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=delta,2005')
- AnnotationURLCitation(end_index=17042, start_index=16911, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=DGC%20elements%20are%20concentrated%20in,In%20%28more')
- AnnotationURLCitation(end_index=17372, start_index=17212, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=like%20the%20DGC%2C%20is%20concentrated,cardiac%20muscle%2C%20titin%2C%20MLP%20and')
- AnnotationURLCitation(end_index=17655, start_index=17524, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=DGC%20elements%20are%20concentrated%20in,In%20%28more')
- AnnotationURLCitation(end_index=17970, start_index=17839, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=DGC%20elements%20are%20concentrated%20in,In%20%28more')
- AnnotationURLCitation(end_index=18423, start_index=18271, title='Sarcoglycans are enriched at the neuromuscular junction in a nerve-dependent manner - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC11754441/#:~:text=Sarcoglycans%20are%20enriched%20at%20the,48.Massopust%20RT%2C%20Lee')
- AnnotationURLCitation(end_index=18997, start_index=18822, title='A Scoping Review of Sarcoglycan Expression in Non-Muscle Organs: Beyond Muscles - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12294094/#:~:text=This%20scoping%20review%20explores%20the,cells%2C%20implicating%20their%20involvement%20in')
- AnnotationURLCitation(end_index=19164, start_index=18998, title='A Scoping Review of Sarcoglycan Expression in Non-Muscle Organs: Beyond Muscles - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12294094/#:~:text=highlights%20the%20widespread%20presence%20of,that%20they%20may%20contribute%20to')
- AnnotationURLCitation(end_index=19527, start_index=19361, title='A Scoping Review of Sarcoglycan Expression in Non-Muscle Organs: Beyond Muscles - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12294094/#:~:text=highlights%20the%20widespread%20presence%20of,These%20findings%20underscore%20the')
- AnnotationURLCitation(end_index=19810, start_index=19683, title='A Scoping Review of Sarcoglycan Expression in Non-Muscle Organs: Beyond Muscles - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12294094/#:~:text=SG%20subunits%20in%20neurons%20and,However')
- AnnotationURLCitation(end_index=20123, start_index=19982, title='A Scoping Review of Sarcoglycan Expression in Non-Muscle Organs: Beyond Muscles - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12294094/#:~:text=structures%2C%20with%20distinct%20regional%20and,However')
- AnnotationURLCitation(end_index=20470, start_index=20320, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=Dystrophy%20and%20referred%20to%20as,has%20important%20roles%20in%20both')
- AnnotationURLCitation(end_index=20555, start_index=20471, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=%CE%B5')
- AnnotationURLCitation(end_index=20903, start_index=20759, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=absent%20cardiac%20phenotype%20associated%20with,sarcoglycan.%2050')
- AnnotationURLCitation(end_index=20989, start_index=20904, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=allele.')
- AnnotationURLCitation(end_index=21388, start_index=21228, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=not%20associated%20with%20any%20muscle,sarcoglycan%20protein%20has%20homology%20to')
- AnnotationURLCitation(end_index=21474, start_index=21389, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=allele.')
- AnnotationURLCitation(end_index=21903, start_index=21744, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=interactions%20within%20the%20sarcoglycan%20complex,this%20may%20be%20a%20feature')
- AnnotationURLCitation(end_index=21989, start_index=21904, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=allele.')
- AnnotationURLCitation(end_index=22261, start_index=22134, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=%CE%B2,29%7D%20Notably%2C%20the%20smooth%20muscle')
- AnnotationURLCitation(end_index=22346, start_index=22262, title='The Sarcoglycans - Madame Curie Bioscience Database - NCBI Bookshelf', type='url_citation', url='https://www.ncbi.nlm.nih.gov/books/NBK6317/#:~:text=%CE%B4')
- AnnotationURLCitation(end_index=22850, start_index=22723, title='A Scoping Review of Sarcoglycan Expression in Non-Muscle Organs: Beyond Muscles - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12294094/#:~:text=SG%20subunits%20in%20neurons%20and,However')
- AnnotationURLCitation(end_index=23697, start_index=23568, title='Loss of sarcolemma nNOS in sarcoglycan-deficient muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/12409321/#:~:text=PubMed%20pubmed,Our%20data%20suggest%20that%20loss')
- AnnotationURLCitation(end_index=24287, start_index=24119, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=integrated%2C%20the%20sarcoglycan%20complex%20plays,been%20established%20to%20study%20the')
- AnnotationURLCitation(end_index=24907, start_index=24740, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=sarcoglycan%20complex%20binds%20other%20components,been%20established%20to%20study%20the')
- AnnotationURLCitation(end_index=25321, start_index=25182, title='Sarcoglycan Alpha Mitigates Neuromuscular Junction Decline in Aged Mice by Stabilizing LRP4 - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6181315/#:~:text=for%20reduced%20LRP4,NMJ%20stability%20in%20aged%20mice')
- AnnotationURLCitation(end_index=25835, start_index=25664, title='Sarcoglycan Alpha Mitigates Neuromuscular Junction Decline in Aged Mice by Stabilizing LRP4 - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6181315/#:~:text=During%20aging%2C%20acetylcholine%20receptor%20,LRP4%20degradation%20as%20a%20mechanism')
- AnnotationURLCitation(end_index=25995, start_index=25836, title='Sarcoglycan Alpha Mitigates Neuromuscular Junction Decline in Aged Mice by Stabilizing LRP4 - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6181315/#:~:text=suggesting%20compromised%20agrin,serves%20as%20a%20pathological%20mechanism')
- AnnotationURLCitation(end_index=26272, start_index=26133, title='Sarcoglycan Alpha Mitigates Neuromuscular Junction Decline in Aged Mice by Stabilizing LRP4 - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6181315/#:~:text=for%20reduced%20LRP4,NMJ%20stability%20in%20aged%20mice')
- AnnotationURLCitation(end_index=26574, start_index=26435, title='Sarcoglycan Alpha Mitigates Neuromuscular Junction Decline in Aged Mice by Stabilizing LRP4 - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6181315/#:~:text=for%20reduced%20LRP4,NMJ%20stability%20in%20aged%20mice')
- AnnotationURLCitation(end_index=27237, start_index=27085, title='Sarcoglycans are enriched at the neuromuscular junction in a nerve-dependent manner - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC11754441/#:~:text=Sarcoglycans%20are%20enriched%20at%20the,48.Massopust%20RT%2C%20Lee')
- AnnotationURLCitation(end_index=28047, start_index=27872, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=The%20sarcoglycan%20genes%20were%20identified,milder%20cases%2C%20in%20which%20patients')
- AnnotationURLCitation(end_index=28520, start_index=28345, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=The%20sarcoglycan%20genes%20were%20identified,milder%20cases%2C%20in%20which%20patients')
- AnnotationURLCitation(end_index=28769, start_index=28610, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=patients%20develop%20progressive%20proximal%20muscle,Melacini%20et%20al')
- AnnotationURLCitation(end_index=29095, start_index=28907, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=sarcoglycanopathies%20are%20severe%20disorders%20characterized,of%20protein%20expression%20cause%20a')
- AnnotationURLCitation(end_index=29511, start_index=29375, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=clinical%20data%20were%20collected%20for,T.%20We')
- AnnotationURLCitation(end_index=29771, start_index=29630, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=clinical%20data%20were%20collected%20for,525delT%20or')
- AnnotationURLCitation(end_index=30099, start_index=29959, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=the%20analysis%20because%20of%20insufficient,T.%20We')
- AnnotationURLCitation(end_index=30562, start_index=30398, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=Sixty%20per%20cent%20of%20LGMDR3,our%20knowledge%20about%20these%20extremely')
- AnnotationURLCitation(end_index=30907, start_index=30753, title='Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/#:~:text=the%20sarcoglycan%20complex%20is%20of,This%20review%20summarises%20the')
- AnnotationURLCitation(end_index=31366, start_index=31190, title='New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy | Brain | Oxford Academic', type='url_citation', url='https://academic.oup.com/brain/article/143/9/2696/5900350#:~:text=relaxation%20through%20association%20with%20the,1998%3B%20Lim%20and%20Campbell%2C%201998')
- AnnotationURLCitation(end_index=32150, start_index=31974, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=Accordingly%2C%20pharmacologic%20and%20genetic%20inhibition,sarcoglycan%E2%80%93null%20mice')
- AnnotationURLCitation(end_index=32305, start_index=32151, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=the%C2%A0eATP%2FP2X%20purinergic%20pathway%20by%20a,sarcoglycanopathy')
- AnnotationURLCitation(end_index=32690, start_index=32536, title='The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of α-Sarcoglycan–Deficient Muscular Dystrophy - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC12179516/#:~:text=the%C2%A0eATP%2FP2X%20purinergic%20pathway%20by%20a,sarcoglycanopathy')
- AnnotationURLCitation(end_index=33245, start_index=33065, title='Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/36289891/#:~:text=strength%20significantly%20improved%20after%20treatment,percentage%20eMyHC%20positive%20fibers%20over')
- AnnotationURLCitation(end_index=33335, start_index=33246, title='Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/36289891/#:~:text=Figure%202')
- AnnotationURLCitation(end_index=33597, start_index=33470, title='Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/36289891/#:~:text=Nintedanib%20reduces%20muscle%20fibrosis%20in,NT')
- AnnotationURLCitation(end_index=33716, start_index=33598, title='Nintedanib Reduces Muscle Fibrosis and Improves Muscle Function of the Alpha-Sarcoglycan-Deficient Mice - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/36289891/#:~:text=shown,Scale%20bar%20%3D%20200%20%CE%BCm')
- AnnotationURLCitation(end_index=34574, start_index=34442, title='LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/#:~:text=Alpha,and%20persistence%20of%20gene%20expression')
- AnnotationURLCitation(end_index=34666, start_index=34575, title='LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/#:~:text=Methods')
- AnnotationURLCitation(end_index=35063, start_index=34972, title='LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/#:~:text=Methods')
- AnnotationURLCitation(end_index=35467, start_index=35376, title='LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/#:~:text=Results')
- AnnotationURLCitation(end_index=35629, start_index=35468, title='LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/#:~:text=No%20adverse%20events%20were%20encountered,capsid%20specific%20T%20cells%20by')
- AnnotationURLCitation(end_index=35944, start_index=35783, title='LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/#:~:text=No%20adverse%20events%20were%20encountered,capsid%20specific%20T%20cells%20by')
- AnnotationURLCitation(end_index=36219, start_index=36084, title='LGMD 2D gene therapy restores alpha-sarcoglycan and associated proteins - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/#:~:text=transfer%20without%20further%20immune%20suppression')
- AnnotationURLCitation(end_index=36522, start_index=36404, title='Sustained alpha-sarcoglycan gene expression following gene transfer in LGMD2D - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC2970162/#:~:text=Persistent%20alpha,This%20was%20in')
- AnnotationURLCitation(end_index=36682, start_index=36523, title='Sustained alpha-sarcoglycan gene expression following gene transfer in LGMD2D - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC2970162/#:~:text=The%20findings%20of%20this%20gene,reversal%20of%20disease%20was%20displayed')
- AnnotationURLCitation(end_index=37007, start_index=36848, title='Sustained alpha-sarcoglycan gene expression following gene transfer in LGMD2D - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC2970162/#:~:text=The%20findings%20of%20this%20gene,reversal%20of%20disease%20was%20displayed')
- AnnotationURLCitation(end_index=37453, start_index=37299, title='Systemic γ-sarcoglycan AAV gene transfer results in dose-dependent correction of muscle deficits in the LGMD 2C/R5 mouse model - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC9929442/#:~:text=correction%20of%20muscle%20deficits%20in,1%7D%2C%20Stephen%20H%20Baine')
- AnnotationURLCitation(end_index=37600, start_index=37454, title='Systemic γ-sarcoglycan AAV gene transfer results in dose-dependent correction of muscle deficits in the LGMD 2C/R5 mouse model - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC9929442/#:~:text=Skip%20to%20main%20content%20Mol,1%7D%2C%20Stephen%20H%20Baine')
- AnnotationURLCitation(end_index=37982, start_index=37860, title='Base editing repairs an SGCA mutation in human primary muscle stem cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC8262330/#:~:text=muscle%20diseases%20has%20not%20yet,90')
- AnnotationURLCitation(end_index=38417, start_index=38280, title='Base editing repairs an SGCA mutation in human primary muscle stem cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC8262330/#:~:text=onset%2C%20severe%2C%20and%20rapidly%20progressive,90')
- AnnotationURLCitation(end_index=38558, start_index=38418, title='Base editing repairs an SGCA mutation in human primary muscle stem cells - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC8262330/#:~:text=affecting%20the%20limbs%2C%20respiratory%20muscles%2C,90')
- AnnotationURLCitation(end_index=39766, start_index=39595, title='Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/#:~:text=through%20the%20cell%27s%20quality%20control,therapy%20of%20these%20genetic%20disorders')
- AnnotationURLCitation(end_index=40109, start_index=39938, title='Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/#:~:text=through%20the%20cell%27s%20quality%20control,therapy%20of%20these%20genetic%20disorders')
- AnnotationURLCitation(end_index=41291, start_index=41152, title='Sarcoglycan Alpha Mitigates Neuromuscular Junction Decline in Aged Mice by Stabilizing LRP4 - PMC', type='url_citation', url='https://pmc.ncbi.nlm.nih.gov/articles/PMC6181315/#:~:text=for%20reduced%20LRP4,NMJ%20stability%20in%20aged%20mice')
- AnnotationURLCitation(end_index=41524, start_index=41357, title='The sarcoglycan complex in skeletal muscle - PubMed', type='url_citation', url='https://pubmed.ncbi.nlm.nih.gov/26709803/#:~:text=sarcoglycan%20complex%20binds%20other%20components,been%20established%20to%20study%20the')
Perplexity
(SGCA-deep-research-perplexity-lite.md)
Perplexity
(SGCA-deep-research-perplexity-lite.md)SGCA (Q16586) Gene: Comprehensive Research Report
Key Concepts and Definitions
SGCA (Sarcoglycan Alpha) is a protein-coding gene in humans that encodes alpha-sarcoglycan, a critical component of the dystrophin-glycoprotein complex (DGC)[1][3][11]. The DGC is a transmembrane protein complex essential for maintaining the structural integrity of muscle fiber membranes by linking the intracellular actin cytoskeleton to the extracellular matrix[1][3][11]. This linkage is vital for muscle cell stability during contraction and relaxation cycles.
Molecular Function:
Alpha-sarcoglycan, the protein product of SGCA, is part of the sarcoglycan subcomplex within the DGC. It contributes to the mechanical stability of the sarcolemma (muscle cell membrane) and is implicated in calcium ion binding, which may play a role in muscle signaling[1][10]. The gene is expressed almost exclusively in striated (skeletal and cardiac) muscle[1].
Biological Processes:
SGCA is involved in muscle organ development, skeletal muscle tissue regeneration, muscle contraction, and response to denervation[10]. Its primary role is to stabilize the muscle membrane and protect muscle fibers from damage during mechanical stress[1][3][11].
Cellular Localization:
Alpha-sarcoglycan localizes to the sarcolemma, specifically within the dystrophin-associated glycoprotein complex and membrane rafts[10]. It is also found at cell-cell junctions and in the cytoplasm, reflecting its role in membrane-cytoskeleton linkage[10].
Protein Domains:
While detailed domain architecture is not fully elucidated in the primary literature, alpha-sarcoglycan is a single-pass transmembrane protein with extracellular, transmembrane, and cytoplasmic regions. It is known to interact with other sarcoglycans (beta, gamma, delta) to form the sarcoglycan complex[1][3][11].
Known Interactions:
SGCA interacts with biglycan, a proteoglycan involved in extracellular matrix organization[3]. It also forms essential interactions within the DGC, particularly with dystrophin, syntrophins, and dystroglycans[3]. These interactions are crucial for the structural and functional integrity of muscle fibers.
Disease Associations:
Mutations in SGCA cause autosomal recessive limb-girdle muscular dystrophy type 2D (LGMDR3), also known as sarcoglycanopathy[1][3][4]. This condition is characterized by progressive muscle wasting, typically beginning in childhood, and often leads to loss of ambulation in adolescence[3][4]. The disease is part of a broader group of muscular dystrophies linked to DGC dysfunction.
Recent Developments and Latest Research (2023–2024)
Gene Editing and Therapeutic Approaches:
Recent studies have demonstrated the potential of base editing to repair SGCA mutations in human primary muscle stem cells, offering a promising avenue for gene therapy in sarcoglycanopathies[5][12]. This approach aims to correct the underlying genetic defect and restore normal protein function, potentially halting or reversing disease progression.
Functional Genomics:
Ongoing research continues to refine the understanding of SGCA’s role within the DGC and its contribution to muscle membrane stability. Advances in proteomics and glycomics are providing deeper insights into post-translational modifications and interactions of alpha-sarcoglycan[10].
Disease Mechanisms:
Studies emphasize that SGCA mutations disrupt the sarcoglycan complex, leading to sarcolemma instability, increased susceptibility to mechanical stress, and eventual muscle fiber necrosis[1][3]. The exact mechanisms by which calcium ion binding by alpha-sarcoglycan contributes to pathology remain an active area of investigation[1][10].
Current Applications and Real-World Implementations
Diagnostics:
Genetic testing for SGCA mutations is a standard part of the diagnostic workup for patients with limb-girdle muscular dystrophy, especially in cases with autosomal recessive inheritance[1][4]. Identification of specific mutations can guide genetic counseling and inform prognosis.
Therapeutic Development:
Gene therapy trials targeting SGCA and other sarcoglycan genes are in preclinical and early clinical phases. Base editing and CRISPR/Cas9 technologies are being explored to correct mutations in patient-derived cells, with the goal of advancing toward clinical trials[5][12]. These approaches represent a significant shift from symptomatic management to potential curative strategies.
Research Tools:
SGCA is used as a model to study membrane-cytoskeleton interactions and the pathophysiology of muscular dystrophies. Cell and animal models with SGCA mutations are employed to test novel therapeutics and understand disease mechanisms[5][12].
Expert Opinions and Analysis
Authoritative Perspectives:
Experts in neuromuscular disorders highlight the centrality of the DGC, and by extension SGCA, in maintaining muscle integrity. Disruption of this complex leads to a cascade of events culminating in muscle degeneration[1][3][11]. The field is moving toward precision medicine, with gene editing offering hope for previously untreatable genetic muscle diseases[5][12].
Critical Challenges:
A major challenge is ensuring the safety and efficacy of gene-editing therapies, particularly with respect to off-target effects and long-term outcomes. Additionally, the heterogeneity of SGCA mutations complicates the development of universal therapies, necessitating personalized approaches[5][12].
Relevant Statistics and Data from Recent Studies
- Prevalence: Limb-girdle muscular dystrophy type 2D is a rare disease, but exact prevalence rates are not well established due to underdiagnosis and genetic heterogeneity.
- Therapeutic Progress: Preclinical studies using base editing in human muscle stem cells have shown correction of SGCA mutations and partial restoration of protein function, though clinical efficacy remains to be demonstrated[5][12].
- Mutation Spectrum: Over 100 SGCA mutations have been reported, most leading to loss of protein function and severe clinical phenotypes[3][4].
Summary Table: SGCA (Q16586) at a Glance
| Aspect | Details |
|---|---|
| Gene Symbol | SGCA |
| Protein Name | Alpha-sarcoglycan |
| UniProt ID | Q16586 |
| Molecular Function | Component of DGC; calcium ion binding; membrane-cytoskeleton linkage |
| Biological Process | Muscle development, regeneration, contraction |
| Cellular Localization | Sarcolemma, membrane rafts, cell-cell junctions |
| Protein Domains | Single-pass transmembrane, extracellular, cytoplasmic regions |
| Key Interactions | Biglycan, other sarcoglycans, dystrophin, syntrophins, dystroglycans |
| Disease Association | Limb-girdle muscular dystrophy type 2D (LGMDR3) |
| Therapeutic Approaches | Gene editing (base editing, CRISPR/Cas9) in preclinical development |
URLs and Publication Dates
- GeneCards: Updated regularly, last accessed November 2025[1].
- UniProt: Entry last modified in 2023[11].
- GlyCosmos: Last updated August 4, 2025[10].
- NCBI Gene: Includes recent studies on base editing (2023–2024)[12].
- Wikipedia: Continuously updated, last substantive edit not specified[3].
Conclusion
SGCA encodes alpha-sarcoglycan, a cornerstone of the dystrophin-glycoprotein complex essential for muscle membrane stability. Mutations in SGCA cause limb-girdle muscular dystrophy type 2D, a progressive and debilitating condition. Recent advances in gene editing offer promising therapeutic avenues, though significant challenges remain in translating these technologies to clinical practice. Ongoing research continues to elucidate the molecular intricacies of SGCA and its role in muscle biology, with the ultimate goal of developing effective treatments for sarcoglycanopathies[1][3][5].
Citations
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=SGCA
- https://patents.google.com/patent/US20230357754A1/en
- https://en.wikipedia.org/wiki/SGCA
- https://g2p.broadinstitute.org/gene/SGCA/protein/Q16586
- https://glygen.org/protein/Q16586
- https://flybase.org/reports/FBgn0032013
- https://functionome.geneontology.org/gene/UniProtKB:Q16586
- http://biogps.org/gene/6442/
- https://marrvel.org/human/gene/6442
- https://glycosmos.org/glycoproteins/Q16586
- https://www.uniprot.org/uniprotkb/Q16586/entry
- https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=6442
- https://www.uniprot.org/uniprot/H0Y8T1_HUMAN
Perplexity
(SGCA-deep-research-perplexity.md)
Perplexity
(SGCA-deep-research-perplexity.md)SGCA Gene: Comprehensive Functional Annotation of Alpha-Sarcoglycan
The SGCA gene encodes alpha-sarcoglycan (α-SG or SGCA), a critical transmembrane glycoprotein component of the dystrophin-glycoprotein complex (DGC) that functions as both a structural protein maintaining muscle fiber membrane integrity and an ATP-hydrolyzing enzyme participating in mechanotransduction at the sarcolemma[1][5][8]. Located on chromosome 17q21, SGCA mutations cause autosomal recessive limb-girdle muscular dystrophy type 2D (LGMD2D/LGMDR3), one of the most common sarcoglycanopathies[3][6][56]. Recent structural studies have revealed that alpha-sarcoglycan participates in a sophisticated tower-like extracellular architecture critical for organizing the entire DGC, establishing its importance not merely as a peripheral component but as a central structural organizer that connects the cytoskeleton to the extracellular matrix through multiple interactive domains[37][40]. This comprehensive report synthesizes current knowledge on SGCA's molecular structure, biochemical functions, subcellular localization, mechanotransductive roles, and disease mechanisms, providing a detailed functional annotation informed by crystallographic structures, biochemical studies, and cell biological investigations.
Molecular Structure and Gene Organization
Genomic Architecture and Expression
The SGCA gene spans the chromosome 17p21 locus and encompasses ten exons that encode a protein of 387 amino acids[3]. The gene structure reflects the organization typical of transmembrane protein genes, with exons distributed across a genomic region that allows for alternative splicing events generating transcript variants encoding different isoforms[1]. Unlike many tissue-specific genes with highly restricted expression patterns, SGCA exhibits relatively confined expression, being primarily restricted to striated muscle tissue including skeletal and cardiac muscle fibers[1][15]. This muscle-specific expression pattern reflects the specialized role of alpha-sarcoglycan in stabilizing the unique membrane architecture of contractile cells. The protein's biosynthesis begins with cotranslational translocation into the endoplasmic reticulum, where the nascent polypeptide undergoes extensive post-translational modifications before trafficking to the plasma membrane[39]. The restriction of SGCA expression to striated muscle tissues distinguishes it from other sarcoglycan family members such as epsilon-sarcoglycan, which displays broader tissue distribution including expression in the central nervous system[32].
Protein Sequence and Structural Domains
Alpha-sarcoglycan is synthesized as a 387-amino acid protein that undergoes signal sequence cleavage and N-linked glycosylation to generate a mature protein of approximately 50 kilodaltons, though the primary amino acid sequence predicts a mass of approximately 43 kDa before glycosylation[5][11][19]. The structural organization of alpha-sarcoglycan comprises several functionally distinct domains including an N-terminal signal sequence, a single transmembrane helix, and an extensive extracellular domain[1][19]. Within the N-terminal extracellular region, alpha-sarcoglycan contains a cadherin-like domain and calcium-binding pockets that are also found in alpha-dystroglycan, suggesting a possible calcium-dependent heterotypic adhesion mechanism between these two proteins[11][19]. The protein sequence analysis reveals five highly conserved cysteine residues within the extracellular portion and two putative N-linked glycosylation sites that are conserved across all species[19]. These cysteine residues are of critical functional importance, as they participate in disulfide bond formation that stabilizes the secondary structure of the protein and are frequently targeted by disease-causing mutations[3][11].
Importantly, alpha-sarcoglycan possesses a putative ATP-binding sequence within its extracellular domain that is conserved across all species[11][19]. This ATP-binding motif confers nucleotide-binding and catalytic capability to the protein, distinguishing alpha-sarcoglycan from the other sarcoglycan subunits. The C-terminus of the protein contains a conserved regulatory region that interacts with other sarcoglycan subunits during complex assembly[14][39]. Analysis of the protein reveals that approximately 5 to 10 percent of its molecular weight derives from N-linked glycosylation, with the major glycan structures attached to consensus sequences within the extracellular domain[11]. The glycosylation process, which begins co-translationally with the attachment of a preformed oligosaccharide (GlcNAc₂Man₉Gluc₃) by oligosaccharyltransferase, is followed by modifications in the endoplasmic reticulum and Golgi apparatus that trim and remodel the glycan structures[39].
Subcellular Localization and Membrane Organization
Sarcolemmal Localization and Costameric Organization
Alpha-sarcoglycan is localized exclusively to the plasma membrane (sarcolemma) of muscle fibers where it functions as a transmembrane component of the dystrophin-glycoprotein complex[1][5]. Within the sarcolemma, alpha-sarcoglycan exhibits a highly organized distribution pattern that reflects its structural role in the complex. Specifically, alpha-sarcoglycan is enriched at costameres, which are sub-membranous cytoskeletal structures aligned with the Z-lines of sarcomeres[20][23]. Costameres serve as specialized mechanical linkage points that transmit lateral forces from the contractile apparatus to the extracellular matrix, and alpha-sarcoglycan's positioning at these sites reflects its critical importance in force transduction[20]. The organization of alpha-sarcoglycan at costameres occurs in strict stoichiometric relationships with other DGC components, with the protein functioning as part of a discrete oligomeric complex rather than as scattered individual molecules[11][19].
Beyond costameres, alpha-sarcoglycan exhibits enrichment at a second specialized membrane domain—the neuromuscular junction (NMJ)[2][60]. Recent detailed immunofluorescence studies have demonstrated for the first time that the entire sarcoglycan complex, including alpha-sarcoglycan, accumulates at postsynaptic regions of the NMJ in an enriched manner[2]. In wild-type muscle fibers, alpha-sarcoglycan signal at the NMJ is colocalized with acetylcholine receptor (AChR) clusters while other sarcoglycan subunits localize adjacent to but not overlapping with AChRs[2]. This specific enrichment at the NMJ requires nerve-derived signals, as denervation experiments demonstrate that the sarcoglycan complex disappears from the NMJ postsynaptic region while persisting in the sarcolemma proper[60]. The differential requirement for nerve signals to establish NMJ-enriched sarcoglycan localization compared to AChR clustering suggests that alpha-sarcoglycan and its associated complex participate in nerve-dependent signaling functions distinct from simple receptive plate formation[2][60].
Biosynthetic Trafficking and Membrane Insertion
The pathway from biosynthesis to membrane localization of alpha-sarcoglycan is remarkably complex and serves as a critical control point for protein function. Nascent alpha-sarcoglycan molecules are cotranslationally inserted into the endoplasmic reticulum where they encounter the ER quality control system[14][39][49]. The protein undergoes extensive folding processes facilitated by molecular chaperones including calnexin and calreticulin, and undergoes oxidation of disulfide bonds through action of protein disulfide isomerases[39]. These folding processes are coupled with N-linked glycosylation events. Following successful ER processing, alpha-sarcoglycan exits the ER at specialized budding sites containing coat protein complex II and enters the Golgi apparatus[39]. Critically, biochemical studies in C2C12 myogenic cells have demonstrated that the simultaneous synthesis of all four sarcoglycans—alpha, beta, gamma, and delta—is mandatory for proper assembly and cell membrane localization of the sarcoglycan complex[14][39]. This requirement for co-synthesis ensures that malformed or incomplete complexes do not assemble or traffic to the cell surface.
The assembly process follows a sequential and well-characterized order of association[14][39]. Beta-sarcoglycan initiates a strong interaction with delta-sarcoglycan, forming a β-δ core that serves as the nucleation site for complex formation. Gamma-sarcoglycan is subsequently added to this β-δ core. Alpha-sarcoglycan is then recruited through interaction primarily with gamma-sarcoglycan, though evidence also suggests possible direct interaction with the β-δ core[14][39]. In primary mouse myotubes, beta-, delta-, and gamma-sarcoglycans form tight interactions, while alpha-sarcoglycan appears less tightly associated, reflecting its more flexible positioning within the complex[14][39]. This differential association strength may relate to the specific role of alpha-sarcoglycan as a terminal subunit incorporating ATP-hydrolysing activity into the complex. Upon reaching the cell membrane, alpha-sarcoglycan does not insert as an isolated molecule but rather as part of the complete tetrameric complex. Notably, beta- and delta-sarcoglycans can be transported to the cell membrane even in the absence of alpha- and gamma-sarcoglycans, suggesting their capacity to form partial complexes; however, normal stoichiometric organization requires all four subunits[14][39].
Primary Structural Functions and Role in the Dystrophin-Glycoprotein Complex
Architecture and Assembly of the Sarcoglycan Subcomplex
The sarcoglycan complex, of which alpha-sarcoglycan is a component, functions as a critical subcomplex within the larger dystrophin-glycoprotein complex that links the intracellular actin cytoskeleton to the extracellular matrix[1][5][7]. The recent high-resolution cryoelectron microscopy (cryoEM) structure of the native DGC isolated from mouse skeletal muscle has revolutionized understanding of alpha-sarcoglycan's structural role, revealing a model markedly divergent from previous biochemical proposals[37][40]. On the extracellular side of the membrane, beta-, gamma-, and delta-sarcoglycans co-fold to form a specialized tower-like structure that serves as the primary binding site for alpha-sarcoglycan[37][40]. This tower-like extracellular architecture provides multiple contact points that stabilize alpha-sarcoglycan's interaction with the complex and appears to have a central role in organizing the entire DGC assembly[37][40]. This novel structural organization explains why mutations in any single sarcoglycan gene result in destabilization and loss of the entire complex from the membrane—the interdependent architecture cannot tolerate loss of any component[3][5][11].
In the transmembrane region, the cryoEM structure reveals that sarcoglycans and sarcospan (a smaller associated protein) flank and stabilize the single transmembrane helix of dystroglycan rather than forming a subcomplex as previously proposed[37][40]. This corrected model indicates that alpha-sarcoglycan participates in an intricate network of interactions spanning the full thickness of the membrane. On the intracellular side, sarcoglycans including alpha-sarcoglycan engage with the dystrophin-dystrobrevin subcomplex through extensive interactions with the ZZ domain of dystrophin[37][40]. These interactions link the sarcoglycan complex to the major structural protein dystrophin, establishing a continuous structural scaffold that transduces mechanical stress across the full membrane thickness[37][40].
Mechanical Stabilization of the Sarcolemma
The primary structural function of alpha-sarcoglycan centers on maintaining mechanical stability of the sarcolemma during the intense forces generated by muscle contraction. The dystrophin-glycoprotein complex, of which alpha-sarcoglycan is a core component, functions as the principal mechanically strong linkage between the sarcolemmal membrane and the costameric γ-actin cytoskeleton[47]. Studies in which inside-out sarcolemmal membranes were mechanically peeled from single muscle fibers demonstrate that the dystrophin complex is necessary for establishing a mechanically strong physical linkage between the sarcolemma and costameric actin[47]. Loss of alpha-sarcoglycan, like loss of other DGC components, compromises this mechanical linkage and destabilizes the entire costameric apparatus[47].
The mechanical requirement for alpha-sarcoglycan is particularly evident during eccentric muscle contractions (lengthening contractions) that generate maximal sarcolemmal stress. During the intense lateral tensions produced by contractile activity, the DGC distributes force across the broad sarcolemmal surface rather than concentrating it at focal adhesion-like structures, protecting the membrane from rupture[7][20]. Dystrophin acts as a molecular spring within this complex, extending up to 84 nanometers under physiological force ranges (15-30 piconewtons) generated by myosin contraction[7][54]. The sarcoglycan complex, including alpha-sarcoglycan, stabilizes this spring-like deformation by maintaining the structural relationships within the complex during force application. When sarcoglycan function is disrupted by mutation, this protective mechanism fails, leading to calcium influx through ruptured membranes, triggering cascades of cellular pathology that result in muscle fiber degeneration[6][11].
Enzymatic Function: ATP-Hydrolysing Activity
Identification and Characterization of ATPase Function
A distinctive feature of alpha-sarcoglycan distinguishing it from other sarcoglycan subunits is its possession of ATP-binding and ATP-hydrolyzing enzymatic activity. Early studies noting the protein's homology to ecto-ATPases prompted investigation of this possibility. Definitive biochemical evidence conclusively demonstrates that alpha-sarcoglycan is a calcium and magnesium-dependent ecto-ATPase that hydrolyzes extracellular ATP and ADP with specificity for these adenine nucleotides[8]. The enzyme activity requires the presence of both Mg²⁺ and Ca²⁺ and is completely inhibited by inhibitors of ecto-ATPases including suramin and reactive blue-2[8]. The substrate specificity of alpha-sarcoglycan is notable—the protein hydrolyzes ATP and ADP but does not hydrolyze other triphosphonucleosides, indicating a substrate specificity restricted to adenine nucleotides[8]. The affinity of alpha-sarcoglycan for ATP is in the low millimolar range, typical of ecto-ATPases that function in environments where nucleotide concentrations can fluctuate substantially[8].
The catalytic properties of alpha-sarcoglycan were definitively characterized through multiple experimental approaches. Correlation of alpha-sarcoglycan protein expression with ecto-nucleotidase activity during differentiation of C2C12 myogenic cells into myotubes demonstrated that approximately 25 percent of the ecto-nucleotidase activity displayed by mature myotubes was inhibited by preincubation with an antibody specific for the ATP-binding motif of alpha-sarcoglycan[8]. Direct demonstration of enzymatic activity was achieved through transfection of human embryonic kidney (HEK) 293 cells with expression plasmids containing alpha-sarcoglycan cDNA, which resulted in significant increases in ATP-hydrolyzing activity that could be completely abolished by addition of anti-alpha-sarcoglycan antibodies[8]. These findings collectively establish that alpha-sarcoglycan substantially contributes to the total ecto-nucleotidase activity of muscle cells and possesses intrinsic enzymatic activity as a Ca²⁺, Mg²⁺-dependent ecto-ATPase.
Functional Implications of ATP Hydrolysis
The physiological role of alpha-sarcoglycan's ATP-hydrolysing activity remains incompletely understood but several mechanisms have been proposed. The ATP-hydrolysing activity occurs on the extracellular face of the protein, positioning it to regulate the levels of extracellular ATP and generate adenosine through sequential hydrolysis of ATP to ADP to adenosine[8][21]. This enzyme activity could modulate purinergic signaling at the muscle cell surface, as extracellular ATP and adenosine serve as signaling molecules that activate P2 purinergic receptors and P1 adenosine receptors respectively on both muscle cells and motor neurons[21][24]. The sequential hydrolysis of ATP to ADP and adenosine creates a temporal profile of purinergic signaling that could coordinate muscle cell responses.
Recent findings demonstrating enrichment of sarcoglycans including alpha-sarcoglycan at the neuromuscular junction have led to hypotheses that the ATP-hydrolyzing activity of alpha-sarcoglycan may participate in neuromuscular transmission signaling[2][60]. Since ATP is released as a cotransmitter with acetylcholine at the neuromuscular junction, and since nerve-dependent signals are required for sarcoglycan enrichment at the NMJ (distinct from AChR clustering requirements), it has been speculated that alpha-sarcoglycan's ability to hydrolyze extracellular ATP at the NMJ may contribute to conformational changes within the sarcoglycan complex that trigger unknown molecular signaling pathways[60]. The fact that this ATP-hydrolysing function requires both calcium and magnesium suggests that it may be further regulated by changes in divalent cation concentrations that accompany neuromuscular transmission. However, direct evidence linking the ATP-hydrolysing activity to specific physiological outcomes remains to be established through further investigation.
Mechanotransduction and Signal Transduction Functions
Role in Mechanical Signal Transduction
Beyond its structural and enzymatic functions, alpha-sarcoglycan and the sarcoglycan complex participate critically in mechanotransduction—the process by which mechanical forces are converted into biochemical signals. Studies examining mechanically activated protein kinase pathways in skeletal muscle have demonstrated that the sarcoglycan complex functions as a mechanoreceptor capable of detecting and transducing mechanical perturbations[13][25]. When muscles experience dynamic mechanical loading such as eccentric contractions, the sarcoglycan complex undergoes rapid phosphorylation events with concomitant activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) in a manner that requires an intact sarcoglycan complex[13][25]. Muscles lacking gamma-sarcoglycan display markedly altered ERK1/2 phosphorylation responses to mechanical stimulation compared to wild-type muscle, with reduced mechanically-induced ERK1 phosphorylation but enhanced ERK2 phosphorylation at rest[25].
The mechanotransductive function of the sarcoglycan complex appears partially distinct from the mechanical stabilization function, as demonstrated by elegant studies using viral-mediated gene transfer to restore either wild-type or functionally impaired gamma-sarcoglycan to gamma-sarcoglycan-deficient muscles[13]. Even when gamma-sarcoglycan containing a critical tyrosine-to-alanine mutation (Y6A) that abolished normal signal transduction was restored to normal membrane localization and fully restored complex assembly, the aberrant signal transduction response associated with gamma-sarcoglycan loss persisted[13]. This dissociation between successful complex localization and restoration of normal signaling responses indicates that specific residues within the sarcoglycan complex are required for transducing mechanical information beyond those necessary for structural integrity and membrane localization. The tyrosine-6 residue within gamma-sarcoglycan appears critical for modulating responses to eccentric contractions, though the precise molecular mechanism by which this tyrosine residue participates in mechanotransduction remains to be elucidated[13].
Integration with Broader Mechanosignaling Pathways
The dystrophin-glycoprotein complex, including alpha-sarcoglycan, integrates mechanotransductive signals through interactions with multiple signaling molecules and pathways. The syntrophins, particularly alpha-1-syntrophin, associate with the DGC through direct interaction with spectrin repeats 16-17 of dystrophin and subsequently interact with neuronal nitric oxide synthase (nNOS)[54]. This physical association localizes nNOS to the sarcolemma where it can produce nitric oxide in response to muscle activation. The sarcoglycan complex may modulate the delivery of mechanical signals to this syntrophin-nNOS module. Additionally, the syntrophins interact with the actin cytoskeleton, potentially allowing the sarcoglycan complex to influence cytoskeletal dynamics in response to mechanical perturbations[54].
The intracellular adaptor proteins rapsyn and dystrobrevin associate with the DGC components and propagate signals from the complex to downstream effectors[44][51]. Dystrobrevin binds to the C-terminus of dystrophin through coiled-coil interactions and simultaneously interacts with syntrophins, creating a molecular scaffold that assembles signaling complexes[51]. The sarcoglycan complex phosphorylation events that occur during eccentric contractions appear to propagate signals through these adaptor proteins to downstream kinase cascades. The precise mechanisms by which specific sarcoglycan phosphorylation events are coupled to ERK1/2 activation and how these events relate to the structural changes induced by mechanical stress remain active areas of investigation.
Assembly, Trafficking, and Quality Control Mechanisms
Coordinated Assembly of the Tetrameric Complex
The assembly of the sarcoglycan complex is a tightly regulated process requiring precise coordination of protein synthesis, folding, and complex formation. The fundamental principle governing sarcoglycan complex assembly is that all four sarcoglycans must be synthesized and assembled together to form a functional complex capable of trafficking to the plasma membrane and inserting into the sarcolemma[14][39][42]. Biochemical analysis in Chinese hamster ovary (CHO) cells definitively demonstrated that the simultaneous synthesis of all four sarcoglycans is absolutely mandatory for proper assembly and cell membrane localization of the sarcoglycan complex[14][39]. When any single sarcoglycan is missing or non-functional, the entire complex fails to assemble and traffic to the membrane; instead, the remaining sarcoglycan subunits are retained in the endoplasmic reticulum and subsequently degraded[14][39].
The molecular basis for this interdependent assembly relates to the structural organization recently revealed by cryoEM studies. The β-δ-sarcoglycan core forms the structural foundation upon which the remaining subunits are organized[37][40]. Beta-sarcoglycan and delta-sarcoglycan contain conserved cysteine residue clusters and specific sequence motifs critical for interaction with gamma-sarcoglycan[35]. These interactions stabilize gamma-sarcoglycan positioning in the complex. Alpha-sarcoglycan then associates through interaction primarily with gamma-sarcoglycan, though evidence supports additional direct interactions with the β-δ core[14][39]. The stoichiometry of the mature complex at the membrane is strictly one copy of each sarcoglycan subunit, though the mechanism that ensures this 1:1:1:1 stoichiometry during assembly remains incompletely understood.
Notably, studies in heterologous cell systems have revealed that partial complexes can sometimes form—beta- and delta-sarcoglycans can insert into membranes even without alpha- and gamma-sarcoglycans[14][39]. However, this occurs only in non-muscle cells with potentially simplified quality control mechanisms. In muscle cells where stringent quality control systems operate, incomplete complexes do not traffic to the membrane. This observation indicates that muscle cells possess specialized quality control mechanisms that distinguish between complete tetrameric sarcoglycan complexes and incomplete assemblies, preventing trafficking of non-functional partial complexes that could compromise membrane integrity.
Endoplasmic Reticulum Quality Control and Degradation
The endoplasmic reticulum quality control (ERQC) system plays a critical role in recognizing properly folded sarcoglycans and permitting their progression to the Golgi apparatus versus identifying misfolded proteins and targeting them for degradation. During ER residence, sarcoglycans including alpha-sarcoglycan undergo N-linked glycosylation through the action of oligosaccharyltransferase, which appends a preformed oligosaccharide composed of three glucose, nine mannose, and two N-acetyl glucosamine residues to asparagine residues within consensus Asn-X-Ser/Thr sequences[39][49]. Following this initial glycosylation, the lectin chaperones calnexin and calreticulin bind to the glycosylated nascent polypeptide and facilitate its folding through ATP-dependent processes[39][49].
Protein folding within the ER is catalyzed by oxidoreductases of the protein disulfide isomerase (PDI) family, which facilitate the formation and isomerization of disulfide bonds critical for maintaining the secondary structure of sarcoglycans[39][49]. The five conserved cysteine residues within the extracellular domain of alpha-sarcoglycan are important substrates for this disulfide bond formation. As sarcoglycan folding progresses, the glycan modifications continue through sequential trimming of glucose residues by glucosidase I and II. At the conclusion of the calnexin-calreticulin binding cycle, the enzyme UDP-glucose glycoprotein glucosyltransferase (UGGT) recognizes imperfectly folded proteins by detecting exposed hydrophobic residues and reattaches a glucose residue to reintroduced the protein back into the calnexin and calreticulin binding cycle[39][49]. Properly folded proteins lacking exposed hydrophobic regions are recognized by UGGT as complete and proceed along the maturation and export pathway[39][49].
If after multiple folding attempts a sarcoglycan remains misfolded—a fate particularly likely for sarcoglycans with destabilizing missense mutations—the protein is targeted for ER-associated degradation (ERAD)[11][39]. ERAD involves retrotranslocation of the misfolded protein from the ER lumen back to the cytosol where it is polyubiquitinated by E3 ubiquitin ligases and degraded by the 26S proteasome[11][39][49]. This strict quality control mechanism prevents trafficking of non-functional sarcoglycans to the plasma membrane where they could compromise the integrity of the DGC complex. However, this quality control system also constitutes a significant pathological mechanism in sarcoglycanopathies, as many missense mutations destabilize sarcoglycan folding just enough to be recognized as misfolded by ERQC without completely abolishing protein function—the mutant protein is degraded despite retaining substantial activity if it could reach the plasma membrane[11][39][55][58].
Genetic Mutations and Molecular Pathogenesis of LGMD2D
Spectrum of SGCA Mutations and Genetic Heterogeneity
Mutations in the SGCA gene cause autosomal recessive limb-girdle muscular dystrophy type 2D (LGMD2D), also designated LGMDR3 according to newer nomenclature[3][56]. Over 78 SGCA variants have been recognized in humans according to the Human Genome Mutation Database, with more than 70 mutations specifically identified in LGMD2D patients[6]. The mutations span the entire gene but display clustering in particular regions, with exon 3 showing unusually high relative frequency of mutations—approximately 50 percent of mutations occur in this single exon[3]. This clustering likely reflects the particular importance of exon 3-encoded sequences for structural stability or complex assembly.
The most common SGCA mutation is a missense substitution at position 229 (CGC→TGC in the DNA sequence), which results in substitution of arginine with cysteine at codon 77 (R77C)[3][6]. This single mutation accounts for up to one-third of SGCA mutations in some populations, indicating its particular frequency in specific genetic backgrounds[6]. The R77C substitution is particularly illustrative of the molecular mechanisms of sarcoglycanopathy pathogenesis, as the mutant alpha-sarcoglycan protein exhibits altered folding that causes it to be recognized as misfolded by the ER quality control system, leading to ERAD and protein degradation despite retaining substantial structural integrity[55][58]. Other frequent mutations include point mutations encoding missense changes that alter critical cysteine residues, many of which are involved in disulfide bond formation and protein stability[3][11].
In addition to point mutations, SGCA mutations encompass compound heterozygous deletions, deletions affecting entire exons, and splice-site mutations that alter splicing patterns[3][16]. A particularly informative case identified a novel missense mutation (c.218 C>T, p.P73L) combined with a partial heterozygous deletion encompassing exons 7 and 8, resulting in complete loss of all four sarcoglycan proteins from the muscle cell surface[3]. This compound heterozygous mutation demonstrates how different mutation types can synergize to cause particularly severe phenotypes through complete disruption of sarcoglycan complex assembly[3].
Molecular Mechanisms of Pathogenesis
The molecular mechanisms by which SGCA mutations cause muscular dystrophy operate through several pathways. For mutations encoding truncated proteins, complete loss of functional protein results in absence of alpha-sarcoglycan from the sarcolemma, which triggers secondary destabilization of the other sarcoglycan subunits due to their interdependent assembly requirements[11][14]. When alpha-sarcoglycan is missing or severely depleted, beta-, gamma-, and delta-sarcoglycans cannot assemble into a complete complex and are also absent from the membrane. This phenomenon—loss of the entire sarcoglycan complex due to dysfunction of a single subunit—explains why mutations in different sarcoglycan genes produce similar muscular dystrophy phenotypes[1][5][11].
For missense mutations that produce amino acid substitutions rather than truncations, the molecular pathogenic mechanism is more complex and relates to altered protein folding[55][58]. Many missense mutations, particularly those affecting cysteine residues critical for disulfide bond formation or residues in predicted secondary structures, produce proteins with subtly altered three-dimensional structure[11]. These misfolded proteins, though potentially retaining substantial biochemical function, are recognized as aberrant by the ER quality control system and are targeted for ERAD degradation rather than being trafficked to the plasma membrane[55][58]. Experimental evidence demonstrates that pharmacological inhibition of ERAD components can rescue such misfolded alpha-sarcoglycan mutants, allowing them to traffic to the membrane where they partially restore complex assembly and function[55][58]. This evidence indicates that for many LGMD2D patients with missense mutations, the molecular defect is not loss of protein function per se but rather aberrant cellular handling of the misfolded protein.
Genotype-Phenotype Correlations and Clinical Heterogeneity
The relationship between specific SGCA mutations and clinical disease severity demonstrates significant heterogeneity. Patients who are homozygous or compound heterozygous for two null mutations (nonsense mutations, frame-shifting deletions, or frame-shifting insertions causing premature termination) uniformly exhibit early-onset disease with rapid progression[3]. These patients typically develop symptoms before age 10 and experience rapid loss of ambulation within two decades, reflecting complete absence of functional alpha-sarcoglycan protein[3][6]. In contrast, patients with missense mutations display substantial clinical variability even when carrying identical mutations, suggesting that genetic background and environmental factors modulate the clinical phenotype[3][45].
Remarkably, even patients bearing the same homozygous missense mutation show dramatically different disease courses[3]. Among four patients with the identical R490Q missense mutation, age of onset varied from 16 to 47 years, muscle involvement patterns differed between Erb type and LGMD type patterns, disease progression rates varied substantially, and histopathology severity scores showed profound differences[45]. This intrafamilial phenotypic heterogeneity among individuals with identical genotypes indicates that factors beyond the primary SGCA mutation—such as genetic background variants in other genes, environmental exposures, or stochastic developmental events—significantly influence disease manifestation[45].
Clinical Manifestations and Tissue Pathology
Primary Clinical Features of LGMD2D
Limb-girdle muscular dystrophy type 2D presents primarily with weakness and atrophy of muscles in the hip, shoulder, and abdominal regions[6][56]. Symptoms often first appear before age 10 years, though in some cases disease manifestation is delayed until adulthood[56]. The pattern of muscle involvement typically begins in the proximal leg and hip muscles, causing difficulties with activities requiring hip strength such as rising from a seated position and climbing stairs. As disease progresses, weakness extends to shoulder girdle muscles, leading to difficulties with overhead activities and progressive functional decline[56]. The disease shows remarkable severity heterogeneity—in some cases symptoms remain mild with no significant effect on lifespan, whereas in extreme cases disease is rapidly progressive and potentially fatal, substantially shortening life expectancy[56]. Importantly, cognitive function and intelligence remain completely unaffected at all disease stages, distinguishing LGMD2D from metabolic or neurological disorders[56].
Muscle Histopathology
Muscle biopsies from LGMD2D patients reveal characteristic features of muscular dystrophy including muscle fiber necrosis, inflammation, and fibrosis. Early in disease progression, muscle fibers display degeneration and regeneration alongside accumulated lipid and connective tissue infiltration. Immunohistochemical staining demonstrates absent or severely reduced alpha-sarcoglycan protein at the sarcolemma of affected muscle fibers, often with simultaneous loss of the other sarcoglycan subunits, reflecting the interdependent assembly requirement[3]. Comparisons of LGMD2D histology with other limb-girdle muscular dystrophies reveal specific features that can aid in differential diagnosis, including variable patterns of fiber regeneration and degeneration and characteristic lobulated fiber morphology[45].
Recent Structural Insights and Contemporary Understanding
High-Resolution CryoEM Structure of the Dystrophin-Glycoprotein Complex
The recent high-resolution cryoelectron microscopy structure of the native DGC isolated from mouse skeletal muscle has fundamentally advanced understanding of alpha-sarcoglycan's structural organization within the complex[37][40]. The structure, published in 2025 in Nature, reveals a previously unappreciated architecture that diverges significantly from earlier biochemical models[37][40]. On the extracellular side of the membrane, beta-, gamma-, and delta-sarcoglycans co-fold in a specialized configuration that forms a tower-like structure extending outward from the membrane[37][40]. This extracellular tower provides the primary binding surface for alpha-sarcoglycan, positioning alpha-sarcoglycan within the assembly such that it contributes to both extracellular and transmembrane interactions[37][40].
In the transmembrane region, the structure reveals that sarcoglycans and sarcospan, rather than forming a separate subcomplex as previously proposed, flank and stabilize the single transmembrane helix of dystroglycan[37][40]. This architectural arrangement means that alpha-sarcoglycan participates in forming a stabilizing scaffold around the dystroglycan transmembrane domain, preventing improper folding or orientation of this critical structural element. On the intracellular side, sarcoglycans engage extensively with the dystrophin-dystrobrevin subcomplex through interactions involving the ZZ domain of dystrophin[37][40]. These intracellular interactions complete the structural bridge spanning the full membrane thickness, linking extracellular matrix-binding components to the intracellular actin cytoskeleton[37][40].
The cryoEM structure provides a foundation for molecular interpretation of many disease-causing mutations. Mutations affecting the tower-like extracellular architecture would be expected to destabilize alpha-sarcoglycan binding and complex assembly, explaining why such mutations cause complete complex loss despite potentially being "missense" mutations affecting single amino acids[37][40]. Mutations in the transmembrane region might disrupt stabilization of dystroglycan, compromising mechanotransduction even if structural integrity is partially preserved[37][40]. Mutations affecting intracellular interfaces might selectively compromise mechanosignaling while partially preserving mechanical stabilization[37][40].
Therapeutic Implications and Future Directions
Current Therapeutic Approaches
Currently, no cure exists for LGMD2D, though multiple therapeutic strategies are under investigation. Gene replacement therapy represents a promising approach, as demonstrated by a clinical trial injecting recombinant adeno-associated virus carrying the human SGCA cDNA into the extensor digitorum brevis muscle of LGMD2D patients[59]. This study achieved sustained SGCA gene expression over three months without severe adverse events, with full sarcoglycan complex restoration in all subjects and increased muscle fiber size in the long-term study subject[59]. However, challenges remain regarding systemic delivery of gene therapy to large muscle groups and managing immune responses to the viral vector.
Pharmacological chaperone approaches represent an alternative strategy targeting the ERAD pathway. Several studies demonstrate that small molecules known as CFTR correctors (compounds originally developed to rescue misfolded cystic fibrosis transmembrane conductance regulator protein) can promote recovery of folding-defective alpha-sarcoglycan mutants, allowing them to bypass ERAD degradation and traffic to the plasma membrane[58]. Treatment with CFTR correctors induces three-to-four-fold increases in R77C-alpha-sarcoglycan protein content in cellular models and primary patient myogenic cells[58]. The rescued protein assembles into functional sarcoglycan complexes that properly localize to the sarcolemma and strengthen muscle fiber membrane resistance to stressful conditions[58]. These findings suggest potential for future clinical development of SGCA-directed pharmacological chaperone therapies.
Base editing, a novel genome editing approach, has successfully corrected SGCA mutations in human primary muscle stem cells with greater than 90 percent correction efficiency[16]. Base editing, which converts cytosine to uracil, was applied to correct an SGCA c.157G>A mutation in patient-derived muscle satellite cells, resulting in restored alpha-sarcoglycan mRNA and protein expression[16]. The corrected cells demonstrated normal myogenic differentiation and crucially contributed to muscle regeneration and repopulation of the satellite cell niche when transplanted into immunocompromised mice[16]. These findings suggest that base editing could eventually provide a curative strategy for certain SGCA mutations if systemic delivery methods can be developed.
Comparative Biology and Evolution
Conservation and Evolutionary Relationships
Alpha-sarcoglycan is highly conserved across vertebrate species, with orthologs identifiable in fish, amphibians, reptiles, mammals, and presumably other vertebrates. The sequence similarity across species, particularly in functionally critical regions such as the ATP-binding motif and cysteine-rich domain, indicates strong selective pressure for conservation of structure and function. Among invertebrate model organisms, sarcoglycan-related sequences have been identified in Caenorhabditis elegans and Drosophila melanogaster, though these organisms possess fewer sarcoglycan genes than mammals[32]. This evolutionary pattern suggests that sarcoglycans arose in early metazoan evolution, with gene duplication events in vertebrates generating the diversity of modern sarcoglycan genes[32].
The relationship between alpha-sarcoglycan and epsilon-sarcoglycan, which share the highest sequence identity among sarcoglycan family members, reflects a vertebrate-specific gene duplication event[32]. Epsilon-sarcoglycan, which is broadly expressed including in the nervous system, appears capable of assuming some functions of alpha-sarcoglycan in certain tissues. In alpha-sarcoglycan null mouse hearts, epsilon-sarcoglycan is upregulated and compensates for the loss of alpha-sarcoglycan, as these mice do not develop cardiomyopathy despite the severe skeletal muscle dystrophy[32]. This functional redundancy between orthologous sarcoglycan subunits provides insights into the essential cellular functions that sarcoglycans perform.
Conclusion
Alpha-sarcoglycan, encoded by the SGCA gene, represents a multifunctional protein integrating structural, enzymatic, and signaling activities critical for skeletal and cardiac muscle integrity. As a component of the dystrophin-glycoprotein complex, alpha-sarcoglycan maintains the mechanical stability of the sarcolemma during intense contractile stresses through its participation in costameric force transduction. The recent high-resolution cryoelectron microscopy structure reveals a sophisticated tower-like extracellular architecture organized by beta-, gamma-, and delta-sarcoglycans that positions alpha-sarcoglycan as a key organizing element of the entire DGC assembly[37][40]. Beyond structural functions, alpha-sarcoglycan possesses intrinsic ATP-hydrolyzing enzymatic activity that likely participates in purinergic signaling, including at the neuromuscular junction where nerve-dependent signals establish sarcoglycan enrichment[2][8][60]. The complex engagement of alpha-sarcoglycan in mechanical signal transduction through ERK1/2 and related pathways indicates roles in sensing and transducing mechanical information critical for adaptation to physiological demands[13][25].
Mutations in SGCA cause LGMD2D through multiple pathogenic mechanisms including loss of protein function and ER-associated degradation of misfolded protein despite retained biochemical function, highlighting the critical importance of both protein folding and structural assembly[11][39][55][58]. The substantial clinical heterogeneity observed even among patients with identical genotypes suggests important roles for genetic background and environmental modifiers in disease manifestation[45]. Emerging therapeutic strategies including gene replacement, pharmacological chaperoning of misfolded proteins, and base editing-mediated genetic correction offer promise for future treatment, though continued investigation of alpha-sarcoglycan's molecular mechanisms will be essential for developing optimized interventions[16][58][59]. Understanding the precise structural basis for alpha-sarcoglycan's multifaceted functions remains an active area of investigation that should yield further insights into both normal muscle physiology and the pathogenesis of sarcoglycanopathies and related muscular dystrophies.
Citations
- https://www.genecards.org/cgi-bin/carddisp.pl?gene=SGCA
- https://www.nature.com/articles/s41419-025-07353-1
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6850699/
- https://www.uniprot.org/uniprotkb/Q16586/entry
- https://www.ncbi.nlm.nih.gov/books/NBK6317/
- https://medlineplus.gov/download/genetics/gene/sgca.pdf
- https://www.nature.com/articles/s42003-022-03980-y
- https://pmc.ncbi.nlm.nih.gov/articles/PMC1133767/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2627791/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8792866/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3279956/
- https://pubs.acs.org/doi/10.1021/bi8014604
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2878052/
- https://www.proteinatlas.org/ENSG00000108823-SGCA
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8262330/
- https://onlinelibrary.wiley.com/doi/10.1111/j.1600-0854.2006.00519.x
- https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=6442
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3770312/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC4536093/
- https://www.uniprot.org/uniprotkb/A0A804HKZ6/entry
- https://www.molbiolcell.org/doi/10.1091/mbc.E20-07-0453
- https://pubmed.ncbi.nlm.nih.gov/29274390/
- https://pubmed.ncbi.nlm.nih.gov/16162659/
- https://pubmed.ncbi.nlm.nih.gov/10893268/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8895749/
- https://core.ac.uk/download/76394888.pdf
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2952238/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC12404760/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC12253981/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8931187/
- https://pubmed.ncbi.nlm.nih.gov/37845322/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6770289/
- https://pubmed.ncbi.nlm.nih.gov/39663450/
- https://www.ahajournals.org/doi/10.1161/circresaha.117.310230
- https://www.rcsb.org/structure/8YT8
- https://pubmed.ncbi.nlm.nih.gov/21294223/
- https://pubmed.ncbi.nlm.nih.gov/9856984/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2834674/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC7931087/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2597960/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC11807010/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC2175263/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6589386/
- https://pubmed.ncbi.nlm.nih.gov/20460376/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC24974/
- https://pubmed.ncbi.nlm.nih.gov/22078962/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC8349188/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC9515174/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5779665/
- https://lgmd2d.org/what-is-lgmd2d%2Fr3%3F
- https://pmc.ncbi.nlm.nih.gov/articles/PMC12294094/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC5886177/
- https://pmc.ncbi.nlm.nih.gov/articles/PMC6014624/
- newer nomenclature[3][56]
- the Human Genome Mutation Database